Descargar la presentación
La descarga está en progreso. Por favor, espere

1
Farmacología Clínica Farmacovigilancia
Dr. Alexis Mejías Delamano Jefe de Trabajos Prácticos Tercera Cátedra de Farmacología Facultad de Medicina U.B.A.

2
Definiciones Farmacología Clínica: Es la parte de la farmacología que utiliza al ser humano como sujeto de experimentación. El instrumento fundamental que es requerido para efectuar la investigación es el estudio clínico. Estudio Clínico: Es cualquier investigación que se realicen en seres humanos con el fin de descubrir los efectos clínicos, farmacológicos u otro efecto farmacodinámico de un producto en investigación así como para estudiar la absorción, metabolismo, distribución y excreción de un producto en investigación, con el objeto de comprobar su eficacia y seguridad. Investigador: Es una persona responsable de la conducción de un estudio clínico en el sitio donde se realiza el estudio. Si un estudio clínico es conducido por un grupo de individuos, el investigador es el líder responsable del grupo (Investigador Principal). Sub-investigador: Cualquier miembro individual del grupo del estudio clínico designado y supervisado por el Investigador Principal para realizar procedimientos críticos relacionados con el estudio.
. Sub-investigador: Cualquier miembro individual del grupo del estudio clínico designado y supervisado por el Investigador Principal para realizar procedimientos críticos relacionados con el estudio.")
3
Definiciones Protocolo: Es un documento en el que se describe los objetivos, diseño, metodología, organización y aspectos estadísticos de un estudio. Aquellas modificaciones del protocolo se denominan enmiendas. Sujetos del estudio: Es un individuo que participa en un estudio clínico ya sea como receptor del producto en investigación o como un sujeto control. Los requisitos para participar en un estudio clínico son: - ser mayor de 21 años - mujeres en edad fértil con un adecuado control de la natalidad - exclusión de las mujeres embarazadas.

4
Definiciones Producto en investigación: Es una forma farmacéutica de un principio activo que se esta probando o usando como referencia en un estudio clínico. Cabe destacar que puede o no estar siendo comercializado en el mercado farmacéutico. Patrocinador: Es un individuo, compañía u organización responsable de iniciar, administrar, controlar y/o financiar un estudio clínico. Algunos de los ejemplos de patrocinadores son los laboratorios multinacionales, investigadores individuales, entes gubernamentales, etc. Buenas Practicas Clínicas (BPC): Es un estándar para el diseño, conducción, realización, monitoreo, auditoria, registro, análisis y reporte de estudios clínicos que proporciona una garantía de que los datos y los resultados reportados son creíbles y precisos y de que están protegidos los derechos, integridad y confidencialidad de los sujetos del estudio.
: Es un estándar para el diseño, conducción, realización, monitoreo, auditoria, registro, análisis y reporte de estudios clínicos que proporciona una garantía de que los datos y los resultados reportados son creíbles y precisos y de que están protegidos los derechos, integridad y confidencialidad de los sujetos del estudio.")
5
Desarrollo de nuevas moléculas farmacológicas
El tiempo promedio en el desarrollo de una nueva molécula ronda los 10 años aproximadamente, desde el descubrimiento o la creación de la molécula hasta la aprobación el fármaco para su comercialización. El costo de todo el proceso de investigación de un fármaco para una determinada patología ronda los millones de dólares aproximadamente.

6
Fases de la Investigación Clínica de nuevos fármacos
Fase 0 (Fase Preclínica) Fase 1 (Administración a Voluntarios Sanos) Fase 2 (Administración en Pacientes) - Temprana o inicial (2a) - Tardía o final (2b) Fase 3 (Estudios Clínicos, doble ciego, controlados, randomizados) Fase 4 (Farmacovigilancia o Estudios de Post-Marketing)
 Fase 1 (Administración a Voluntarios Sanos) Fase 2 (Administración en Pacientes) - Temprana o inicial (2a) - Tardía o final (2b) Fase 3 (Estudios Clínicos, doble ciego, controlados, randomizados) Fase 4 (Farmacovigilancia o Estudios de Post-Marketing)")
7
Fase Preclínica Estudios a realizar:
Toxicidad aguda, sub-aguda y crónica Estudios especiales de toxicidad Carcinogénesis Mutagénesis Reproducción y Desarrollo fetal Estudios de Tolerancia Local Estimación de: índices de seguridad (IS) e índice terapéutico (IT) (Relación entre toxicidad y Eficacia)
 e índice terapéutico (IT) (Relación entre toxicidad y Eficacia)")
8
Estudios de Toxicidad Estudios de Toxicidad Aguda: Son estudios realizados en por lo menos tres especies, de las cuales una deberá ser no roedora. Deben utilizarse al menos dos vías de administración, una de las cuales estará relacionada con la futura utilización terapéutica y la otra deberá asegurar la absorción del fármaco Estudios de toxicidad sub-aguda a dosis repetidas: Deberán realizarse en al menos dos especies, una de las cuales deberá ser no roedora. La duración deberá ser de 12 a 24 semanas de acuerdo a la naturaleza del producto, empleo terapéutico propuesto y especie animal utilizada. La vía de administración deberá estar en relación con el empleo terapéutico propuesto y especie animal utilizada. Toxicidad Crónica: Se deberá utilizar al menos dos especies, una de las cuales será no roedora. La duración no será menor de 24 semanas de acuerdo a la naturaleza del producto, empleo terapéutico propuesto y especie animal utilizada. La vía de administración deberá estar en relación al empleo terapéutico propuesto. Se deberán utilizar no menos de tres dosis, la mayor deberá producir efectos tóxicos demostrables y la menor deberá relacionarse con las dosis terapéutica propuesta. Se consignará la aparición de efectos tóxicos, relación dosis-efecto, la morbilidad, mortalidad, los parámetros bioquímicos, hematológicos y de nutrición, la dosis toxica estimada y la toxicidad especial.

9
Estudios de Toxicidad Teratogénesis: estudios para evaluar los efectos potencialmente embriotóxicos, de toxicidad sobre la madre. Fertilidad: se investiga efectos sobre las funciones gonadales, del ciclo sexual, la frecuencia de la implantación y concepción Periodos perinatales y postnatales: la droga se administra desde el ultimo periodo del embarazo y durante la lactancia. Mutagénesis-Carcinogénesis: Se evalúa a través de la administración crónica de la droga a los animales de experimentación durante la mayor parte de su vida. Estos estudios requieren una gran cantidad de animales de experimentación y además no se obtienen resultados confiables. Se prefieren estudios In Vitro. Tolerancia local: son estudios para detectar fenómenos de irritación y/o necrosis de la piel en el sitio de la aplicación o inyección de un fármaco. Se basa en la observación de la zona durante un tiempo adecuado y la obtención de material para su posterior estudio anatopatológico. Estos estudios son útiles para aquellos fármacos que se van a administrar en forma parenteral o local (tópica)
")
10
Índice de Seguridad e Índice Terapéutico
IS= DL Valor ideal: mayor o igual a 2 DE99 DL1 = Dosis Letal 1 DE99 = Dosis Efectiva 99 DL50 = Dosis Letal 50 DE50 = Dosis Efectiva 50 IT= DL Valor ideal: mayor o igual a 10 DE50

11
Índices de Seguridad e Índice Terapéutico

12
FASE 1 Administración al voluntario sano
Es la primera administración del fármaco en el hombre (voluntario adulto sano) Incluye 20 a 50 personas sanas que son admitidos en una Unidad de Farmacología Clínica donde se realiza el estudio. Se estudia su perfil farmacocinético: porcentaje de absorción, latencia de absorción, vida media de eliminación, Cmax, Tmax, ABC (Área Bajo la Curva), unión proteica, biodisponibilidad absoluta, Volumen de Distribución, etc. Se realizan estudios de dosis únicas, estudio de dosis repetidas, dosis única hasta el máximo preestablecido Evaluación de parámetros clínicos habituales (Examen físico diario, presión arterial, pulso arterial, registros de electrocardiogramas y electroencefalogramas, exámenes de laboratorio para detectar toxicidad hepática, renal, hematológica) Detectar reacciones adversas Se determinan los efectos farmacológicos
 Incluye 20 a 50 personas sanas que son admitidos en una Unidad de Farmacología Clínica donde se realiza el estudio. Se estudia su perfil farmacocinético: porcentaje de absorción, latencia de absorción, vida media de eliminación, Cmax, Tmax, ABC (Área Bajo la Curva), unión proteica, biodisponibilidad absoluta, Volumen de Distribución, etc. Se realizan estudios de dosis únicas, estudio de dosis repetidas, dosis única hasta el máximo preestablecido. Evaluación de parámetros clínicos habituales (Examen físico diario, presión arterial, pulso arterial, registros de electrocardiogramas y electroencefalogramas, exámenes de laboratorio para detectar toxicidad hepática, renal, hematológica) Detectar reacciones adversas. Se determinan los efectos farmacológicos.")
13
FASE 2 (Temprana o Inicial)
Es la primera administración del fármaco en pacientes (20 a 50 pacientes) Se determinan los parámetros farmacocinéticos en los pacientes (absorción, metabolismo, excreción del fármaco). Se evalúan los efectos terapéuticos. Se evalúan los rangos de dosis terapéuticas. Detectar reacciones adversas.
 Se determinan los parámetros farmacocinéticos en los pacientes (absorción, metabolismo, excreción del fármaco). Se evalúan los efectos terapéuticos. Se evalúan los rangos de dosis terapéuticas. Detectar reacciones adversas.")
14
FASE (Tardía o Final) Se basa en estudios clínicos doble ciego, randomizados, controlados contra placebo o comparadores activos en 200 a 500 pacientes. Se evalúan la eficacia terapéutica en dichos pacientes, por ejemplo: en el estudio de un fármaco para la hipertensión arterial su eficacia para disminuir los valores de tensión arterial. Completar los estudios farmacocinéticos. Elección de la dosis definitiva. Detectar reacciones adversas.

15
Fase 3 Ensayo Clínico Corresponden a los estudios clínicos doble ciego, randomizados, multicéntricos, controlados con placebo o comparador activo que comprenden una muestra de a pacientes. Se evalúa la eficiencia terapéutica de los fármacos en estudio. Puede tener una duración prolongada en el caso de enfermedades crónicas o con frecuentes recidivas (2 a 5 años en promedio) Uno de los objetivos es detectar reacciones adversas. Una vez que se finaliza esta fase, cuando existen resultados clínicos positivos, comienza el proceso de aprobación del producto farmacéutico para su comercialización del fármaco para la indicación estudiada.
 Uno de los objetivos es detectar reacciones adversas. Una vez que se finaliza esta fase, cuando existen resultados clínicos positivos, comienza el proceso de aprobación del producto farmacéutico para su comercialización del fármaco para la indicación estudiada.")
16
FASE 4 (Farmacovigilancia y Estudios de Post-Marketing)
Las tareas de farmacovigilancia se realizan después que el medicamento tiene la aprobación para su comercialización por parte de la autoridad regulatoria. Las evaluaciones farmacológicas se realizan en toda la población de pacientes tratados para la patología para la cual fue aprobado el fármaco. Se evalúan nuevas indicaciones, dosis, vías de administración, formas farmacéuticas, etc. Detectar reacciones adversas muy poco frecuentes o generados como consecuencia de su prolongada utilización (cáncer, enfermedades autoinmunes, etc.) Interacciones con medicamentos anteriormente desconocidas. Evalúa resultados a largo plazo, en otras palabras la efectividad del fármaco. Evaluaciones farmacoeconómicas (costo-efectividad, costo-beneficio, costo-utilidad).
 Interacciones con medicamentos anteriormente desconocidas. Evalúa resultados a largo plazo, en otras palabras la efectividad del fármaco. Evaluaciones farmacoeconómicas (costo-efectividad, costo-beneficio, costo-utilidad).")
17
Características de los ensayos clínicos
Doble ciego: es la condición en la cual tanto el paciente como el medico investigador desconocen la medicación que toma cada paciente que interviene en el ensayo clínico. Simple ciego: es la situación en la cual el investigador o el sujeto de la investigación conocen el tratamiento administrado. Abierto: es la situación en la cual el tratamiento administrado es conocido por el investigador y el sujeto de la investigación Multicéntrico: significa que el mismo estudio clínico (con el mismo protocolo) se lleva a cabo en diferentes países del mundo, al mismo tiempo, conducido por diversos investigadores calificados. Controlado: en el caso que el estudio involucre un nuevo medicamento, se comparan los resultados con respecto a otro fármaco utilizado en forma habitual para la patología en estudio o en el caso de ausencia de tratamiento standard para esa enfermedad hasta ese momento, la comparación se lleva a cabo contra placebo. Randomizado: es el proceso de selección basado en el azar de los pacientes en cada una de las ramas de tratamiento en un ensayo clínico. Se basa en el Principio de Equidad que consiste en la asignación de los pacientes a los distintos tratamientos sea establecido por el azar y no por la decisión subjetiva del investigador o el equipo del Centro de Investigación.
 se lleva a cabo en diferentes países del mundo, al mismo tiempo, conducido por diversos investigadores calificados. Controlado: en el caso que el estudio involucre un nuevo medicamento, se comparan los resultados con respecto a otro fármaco utilizado en forma habitual para la patología en estudio o en el caso de ausencia de tratamiento standard para esa enfermedad hasta ese momento, la comparación se lleva a cabo contra placebo. Randomizado: es el proceso de selección basado en el azar de los pacientes en cada una de las ramas de tratamiento en un ensayo clínico. Se basa en el Principio de Equidad que consiste en la asignación de los pacientes a los distintos tratamientos sea establecido por el azar y no por la decisión subjetiva del investigador o el equipo del Centro de Investigación.")
18
¿Cuándo se utiliza Placebo en los ensayos clínicos?
El placebo es una sustancia inerte que se emplea como sustituto de un supuesto medicamento. El placebo carece de efectos farmacológicos per se. Sin embargo puede producir una respuesta biológica debido a los efectos de sugestión asociados a su administración. El efecto nocebo es el efecto indeseable secundario a la administración de placebo a un paciente. El placebo puede ser utilizado como control solo en aquellos casos en que se carece de algún tratamiento especifico aprobado y debidamente demostrado como eficaz y seguro (Tratamiento Standard). Cuando existe un fármaco de probada eficacia y seguridad para el tratamiento de la afección (tratamiento “gold standard” o de primera elección). Si se pone en peligro la vida o la capacidad física o mental del paciente o los síntomas se tornan intolerables. En estos casos se puede utilizar medicación de rescate y “romper el ciego”.
. Cuando existe un fármaco de probada eficacia y seguridad para el tratamiento de la afección (tratamiento gold standard o de primera elección). Si se pone en peligro la vida o la capacidad física o mental del paciente o los síntomas se tornan intolerables. En estos casos se puede utilizar medicación de rescate y romper el ciego .")
19
Investigación Clínica y Ética
La investigación clínica se realiza solo si se cumplen los requerimientos éticos establecidos en las normativas nacionales e internacionales (Disposición 6677/10 de ANMAT, Good Clinical Practices (GCP), ICH). Comités de Ética: Es un grupo independiente del estudio constituido por profesionales médicos y no médicos (enfermeras, farmacéuticos, abogados, arquitectos, etc.) y además por no profesionales (amas de casa, religiosos, comerciantes, etc.). El Comité de Ética asignado a cada estudio tiene la función de: - verificar que se protejan la seguridad, integridad, bienestar y los derechos de los participantes en el estudio. - considera los aspectos éticos del estudio y los documentos correspondientes (Protocolos, consentimiento informado, información que se le proporcione a los pacientes, procedimientos de reclutamiento, etc.). - realiza revisiones periódicas del estudio.
, ICH). Comités de Ética: Es un grupo independiente del estudio constituido por profesionales médicos y no médicos (enfermeras, farmacéuticos, abogados, arquitectos, etc.) y además por no profesionales (amas de casa, religiosos, comerciantes, etc.). El Comité de Ética asignado a cada estudio tiene la función de: - verificar que se protejan la seguridad, integridad, bienestar y los derechos de los participantes en el estudio. - considera los aspectos éticos del estudio y los documentos correspondientes (Protocolos, consentimiento informado, información que se le proporcione a los pacientes, procedimientos de reclutamiento, etc.). - realiza revisiones periódicas del estudio.")
20
Consentimiento Informado
Es un documento que confirma la participación voluntaria de las personas en un estudio clínico en particular. Solamente puede ser obtenido después que se le haya explicado ampliamente al paciente todos los aspectos del estudio en el que participaría (objetivos del estudio clínico, potenciales beneficios y/o riesgos o inconvenientes de los distintos tratamientos, los tratamientos alternativos disponibles, la confidencialidad de los datos del paciente, los responsabilidades del paciente, médico y otros profesionales de la salud involucrados, etc.). El consentimiento informado debe ser firmado por el paciente o su representante legal en el caso que este no estuviese capacitado para hacerlo (niños, ancianos, pacientes psiquiátricos, incapacitados civiles, etc.), por un testigo (generalmente un familiar) y por último por el investigador que informó al paciente sobre todos los aspectos del estudio clínico. El paciente debe recibir una copia del consentimiento informado firmado y fechado, así como también la información concerniente acerca del estudio. Otra copia del consentimiento informado es adjuntada en la Historia Clínica del paciente en poder del medico tratante, quien debe ser informado de su participación.
. El consentimiento informado debe ser firmado por el paciente o su representante legal en el caso que este no estuviese capacitado para hacerlo (niños, ancianos, pacientes psiquiátricos, incapacitados civiles, etc.), por un testigo (generalmente un familiar) y por último por el investigador que informó al paciente sobre todos los aspectos del estudio clínico. El paciente debe recibir una copia del consentimiento informado firmado y fechado, así como también la información concerniente acerca del estudio. Otra copia del consentimiento informado es adjuntada en la Historia Clínica del paciente en poder del medico tratante, quien debe ser informado de su participación.")
21
Consentimiento Informado
El consentimiento informado de un estudio clínico remarca los siguientes aspectos: la participación voluntaria del paciente en el estudio la confidencialidad de sus datos y registros médicos la duración aproximada de su participación en el estudio y la cantidad de participantes que incluye las personas relacionadas al estudio a contactar ante cualquier eventualidad sus responsabilidades como participante del ensayo los distintos tratamientos y la posibilidad de asignación aleatoria a cada uno de ellos

22
A.N.M.A.T. ¿Qué es la A.N.M.A.T.? Es la Administración Nacional de Medicamentos, Alimentos y Tecnología Medica. Regula todos los procesos pertinentes a la fabricación, distribución, comercialización, promoción e investigación de medicamentos en la Republica Argentina. La principal norma regulatoria del país es la Disposición 6677 del año 2010 de ANMAT. Establece las normativas obligatorias que regula la realización de los ensayos clínicos en el país. Contempla los aspectos relacionados a la ética aplicada a la investigación, la fidelidad de los datos obtenidos, las obligaciones de los investigadores y los patrocinadores, los derechos de los pacientes, el monitoreo del estudio, etc. Good Clinical Practices (GCP): establecidas como una norma para la realización de los ensayos clínicos en humanos en todo el mundo.
: establecidas como una norma para la realización de los ensayos clínicos en humanos en todo el mundo.")
23
Drogas Huérfanas (Orfan Drugs)
Una droga huérfana esta definida como aquella molécula que es útil para el tratamiento de enfermedades de baja prevalencia (Rare Disease). Una enfermedad se considera rara o de baja prevalencia cuando esta presente en menos de 1 cada personas o posee una prevalencia menor al 0.5 – 1 %. Se caracterizan por tener un alto valor para la comunidad médica, se pueden utilizar para otras enfermedades comunes, se pueden investigar otras indicaciones adicionales. Una dificultad es la cantidad reducida de pacientes plausibles de participar en un ensayo clínico.
. Una enfermedad se considera rara o de baja prevalencia cuando esta presente en menos de 1 cada personas o posee una prevalencia menor al 0.5 – 1 %. Se caracterizan por tener un alto valor para la comunidad médica, se pueden utilizar para otras enfermedades comunes, se pueden investigar otras indicaciones adicionales. Una dificultad es la cantidad reducida de pacientes plausibles de participar en un ensayo clínico.")
24
Farmacovigilancia La Farmacovigilancia es la ciencia y las actividades relacionadas con la detección, evaluación, conocimiento y prevención de reacciones adversas y otros posibles problemas relacionados con los medicamentos. Es el conjunto de métodos y observaciones que permiten durante el periodo de comercialización o uso extendido de un medicamento, detectar las reacciones adversas y efectos terapéuticos no previstos en las etapas previas de estudio. El objetivo de la Farmacovigilancia es evaluar permanentemente los efectos adversos observados tras la administración tanto de medicamentos vendidos con receta como aquellos expendidos en forma libre (OTC), a través de la identificación y cuantificación del riesgo. Recientemente, las incumbencias de la farmacovigilancia han sido extendidas e incluye: hierbas, medicamentos tradicionales y complementarios, productos hemoderivados y biológicos, vacunas y dispositivos médicos (tecnovigilancia). Se realiza durante todo el ciclo de vida de un fármaco, tanto en la etapa de investigación como en la etapa Post-Marketing (lanzamiento, crecimiento, estabilización, declinación o reposicionamiento).
, a través de la identificación y cuantificación del riesgo. Recientemente, las incumbencias de la farmacovigilancia han sido extendidas e incluye: hierbas, medicamentos tradicionales y complementarios, productos hemoderivados y biológicos, vacunas y dispositivos médicos (tecnovigilancia). Se realiza durante todo el ciclo de vida de un fármaco, tanto en la etapa de investigación como en la etapa Post-Marketing (lanzamiento, crecimiento, estabilización, declinación o reposicionamiento).")
25
Farmacovigilancia “Todas las sustancias son venenos, no existe ninguna que no lo sea. La dosis diferencia un veneno de un remedio”. PARACELSO ( )
")
26
Farmacovigilancia Evento adverso
Las reacciones adversas comprenden: signos, síntomas, síndromes (conjunto de signos y síntomas), alteraciones en los valores de laboratorio, aparición de una patología nueva, malformaciones fetales, etc. Definición según GCP-ICH (Good Clinical Practices – International Conference of Harmanization) se puede definir un evento adverso como: Una reacción adversa es cualquier manifestación nociva, clínica o biológica imputable a un medicamento o especialidad medicinal, que ocurre a las dosis habitualmente utilizada en el ser humano para la profilaxis, diagnóstico o tratamiento de una enfermedad. “Cualquier ocurrencia medica adversa en un paciente o sujeto de una investigación clínica a quien se le administra un producto medicinal farmacéutico y que no necesariamente tiene una relación causal con este tratamiento. Por lo tanto, un evento adverso puede ser cualquier signo desfavorable y no intencionado (incluyendo un hallazgo anormal de laboratorio), síntoma o enfermedad asociada temporalmente con el uso de un producto medicinal (de investigación) este o no relacionado”.
, alteraciones en los valores de laboratorio, aparición de una patología nueva, malformaciones fetales, etc. Definición según GCP-ICH (Good Clinical Practices – International Conference of Harmanization) se puede definir un evento adverso como: Una reacción adversa es cualquier manifestación nociva, clínica o biológica imputable a un medicamento o especialidad medicinal, que ocurre a las dosis habitualmente utilizada en el ser humano para la profilaxis, diagnóstico o tratamiento de una enfermedad. Cualquier ocurrencia medica adversa en un paciente o sujeto de una investigación clínica a quien se le administra un producto medicinal farmacéutico y que no necesariamente tiene una relación causal con este tratamiento. Por lo tanto, un evento adverso puede ser cualquier signo desfavorable y no intencionado (incluyendo un hallazgo anormal de laboratorio), síntoma o enfermedad asociada temporalmente con el uso de un producto medicinal (de investigación) este o no relacionado .")
27
Farmacovigilancia en Argentina
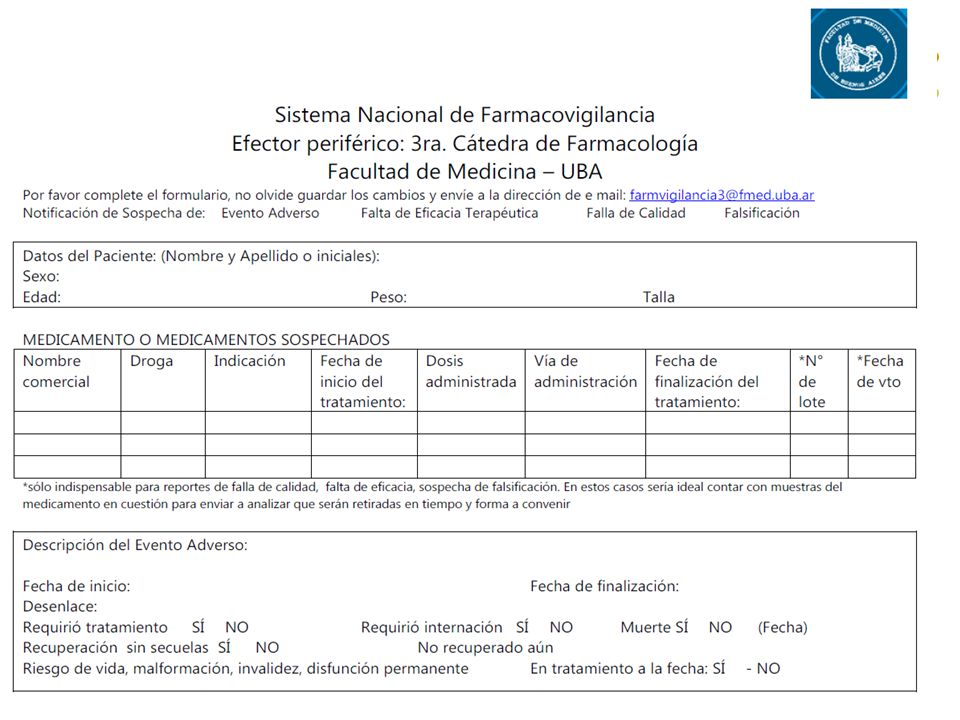
En Argentina, la Farmacovigilancia se basa en las notificaciones realizadas por los profesionales de la salud, las cuales se caracterizan por ser espontáneas y confidenciales, las notificaciones de los pacientes y/o consumidores así como también por las notificaciones realizadas periódicamente por los laboratorios productores de medicamentos (Periodic Safety Reports - PSURs). Las notificaciones pueden ser realizadas por cualquiera de los profesionales de la salud (médicos, enfermeras, farmacéuticos, odontólogos, etc.) y por la población en general (servicio de consumidores, pacientes, etc.).
. Las notificaciones pueden ser realizadas por cualquiera de los profesionales de la salud (médicos, enfermeras, farmacéuticos, odontólogos, etc.) y por la población en general (servicio de consumidores, pacientes, etc.).")
28
Formas de comunicación de las Reacciones Adversas
Pagina web de A.N.M.A.T. Telefónicamente al / Correo postal (envió de la hoja amarilla) Personalmente en A.N.M.A.T. Avenida de Mayo 869, Ciudad Autónoma de Buenos Aires. Efectores periféricos (Hospitales, Cátedras de Farmacología, Centros de Toxicología, Ministerios de Salud provinciales). Departamento de Farmacovigilancia de los laboratorios productores de las especialidades medicinales (Dirección Médica/Asuntos Médicos).
 Personalmente en A.N.M.A.T. Avenida de Mayo 869, Ciudad Autónoma de Buenos Aires. Efectores periféricos (Hospitales, Cátedras de Farmacología, Centros de Toxicología, Ministerios de Salud provinciales). Departamento de Farmacovigilancia de los laboratorios productores de las especialidades medicinales (Dirección Médica/Asuntos Médicos).")
31
Clasificación de los eventos adversos
Relación causal: Se trata de determinar si el evento adverso se puede imputar al fármaco o a sus excipientes. Para determinar la relación causal de un efecto adverso se utilizan criterios basados en la información obtenida a través de la notificación espontánea, estudios prospectivos farmacoepidemiológicos, reportes de casos, notificaciones de estudios clínicos en curso, etc.

32
Criterios de imputabilidad de un evento adverso a un fármaco
1) Relación temporal entre la administración del fármaco y la aparición del signo o síntoma. 2) El síntoma o signo reaparece cuando se vuelve a administrar la droga o se aumenta la dosis de esta. 3) El síntoma o signo desaparece cuando se suspende la administración de la droga o cuando se disminuye la dosis. 4) El signo o síntoma no puede ser explicado por la enfermedad del paciente, enfermedades asociadas o algún tratamiento o droga. 5) El signo o síntoma ha sido reportado anteriormente como asociado al uso de este fármaco.
 Relación temporal entre la administración del fármaco y la aparición del signo o síntoma. 2) El síntoma o signo reaparece cuando se vuelve a administrar la droga o se aumenta la dosis de esta. 3) El síntoma o signo desaparece cuando se suspende la administración de la droga o cuando se disminuye la dosis. 4) El signo o síntoma no puede ser explicado por la enfermedad del paciente, enfermedades asociadas o algún tratamiento o droga. 5) El signo o síntoma ha sido reportado anteriormente como asociado al uso de este fármaco.")
33
Clasificación de las Reacciones Adversas
Reacción adversa definida: Criterios 1,2,3,4,5 Reacción adversa probable: Criterios 1,3,4, 5 Reacción adversa posible: Criterios 1, 3, 5 Reacción adversa condicional: Criterios 1 y 3 Reacción adversa dudosa: el evento adverso esta relacionado con otros factores y no con la droga.

34
Reacciones Adversas Clasificación según la relación causal con el fármaco
Reacción adversa: muestra una relación temporal razonable después de la administración del medicamento, muestra un patrón de respuesta que se conoce se asocia con el medicamento en cuestión, no se explica por la enfermedad del paciente y mejora al suspender el tratamiento y aparece al readministrar el fármaco o al aumentar la dosis. Probable: muestra una relación temporal razonable después de la administración del medicamento, muestra un patrón de respuesta conocido, se confirma después de la suspensión del medicamento (pero no se ha vuelto a administrar) y no se puede explicar por las características de la enfermedad. Posible: muestra una relación temporal razonable, la sintomatología ha sido reportada como reacción adversa asociada al fármaco en consideración y revierte al suspender o disminuir la dosis del fármaco, aunque se puede explicar también por las características propias de la enfermedad o por otros tratamientos concomitantes. Condicional: muestra una relación temporal razonable, pero no existe reporte previo. La sintomatología revierte al reducir o suspender el fármaco pero puede explicarse por otra droga o condición del paciente. Dudosa: no cumple con ninguno de los criterios anteriores, el evento esta relacionado, probablemente, a otros factores.
 y no se puede explicar por las características de la enfermedad. Posible: muestra una relación temporal razonable, la sintomatología ha sido reportada como reacción adversa asociada al fármaco en consideración y revierte al suspender o disminuir la dosis del fármaco, aunque se puede explicar también por las características propias de la enfermedad o por otros tratamientos concomitantes. Condicional: muestra una relación temporal razonable, pero no existe reporte previo. La sintomatología revierte al reducir o suspender el fármaco pero puede explicarse por otra droga o condición del paciente. Dudosa: no cumple con ninguno de los criterios anteriores, el evento esta relacionado, probablemente, a otros factores.")
35
Reacciones Adversas Clasificación según severidad
Las reacciones adversas serias son todas aquellas situaciones u ocurrencias medicas desfavorables que pueden provocar: muerte amenaza a la vida hospitalización del paciente o prolongación de una hospitalización previa. incapacidad persistente o significativa. cáncer anomalías fetales / defectos de nacimiento

36
Tipos de Reacciones Adversas
Efecto Nocebo: Es el efecto adverso producido tras la administración de placebo a los pacientes, generalmente en el ámbito de un ensayo clínico. Efectos colaterales: se observan con dosis terapéuticas de los fármacos. Son la consecuencia de la amplia distribución de un receptor y la acción de una droga sobre mas de un receptor, o sea que el receptor no se encuentra solamente en el órgano blanco del tratamiento. Idiosincrasia: Son efectos adversos de baja frecuencia y de mecanismo desconocido. No esta relacionado a las dosis. Síndrome de supresión: Son consecuencia de la supresión o suspensión brusca de la administración crónica de un fármaco. Ejemplo: glucocorticoides, benzodiazepinas.

37
Gracias por su atención!!

Presentaciones similares










![M K Unnikrishnan [Agosto 2006]](/3/1105489/big_thumb.jpg "M K Unnikrishnan [Agosto 2006]>")

 ¿Qué mide? La potencia de las drogas determinando la concentración.>")



>")



