Descargar la presentación
La descarga está en progreso. Por favor, espere

1
Efecto de Buprenorfina en pancreatitis aguda y crónica
Introducción Efecto de Buprenorfina en pancreatitis aguda y crónica UNIDAD GASTROENTEROLOGIA, DEPARTAMENTO MEDICINA INTERNA, FACULTAD MEDICINA , UNVERSIDAD CONCEPCIÓN UNIDAD GASTROENTEROLOGIA, HOSPITAL TRABAJADOR CONCEPCION GASTROENTEROLOGIA, MEDICINA INTERNA, HOSPITAL CLINCO REGIONAL CONCEPCION UNIDAD BIOLOGÍA CELULAR, MLECULAR Y CITOMETRIA FLUJO GRUNENTAL, CHILE CENTRO ESTUDIOS LATINOAMERICANOS DEL CANCER (CELDEC) CENTRO GASTROENTEROLOGICO LOS ARRAYANES (ONG) CLUB DEL PANCREAS: . DRA CECILIA CASTILLO. CLINICA ALEMANA STAGO . DR ZOLTAN BERGER. CLINICA ALEMANA . DR RODRIGO PONCE. INSTITUTO CHILENO JAPONES DEL CANCER . HOSP SAN BORJA ARRIARÁN DR FERNANDO KAWAGUCHI(1, 2) DR RODRIGO LOAIZA(1,3) DR FERNANDO RIQUELME(1,3) DR CARLOS BRICEÑO(1,2) DR GONZALO ZULOAGA (1,3) DR GERMAN ABRIGÓ(1,3) DR PATRICIO TORRES (1 ) MG JL CASTILLO (4) AL CRISTIAN VASQUEZ PARRA (1) AL MARCOS AVILA (5) AL CYNTHIA PEREZ (1) ING. 3D ALEJANDRO SEPULVEDA(6,7) COORD LILIAN SOTO(6,7) NUTRIC. BLANCA BOERO(7) SICOL. LISETTE RIQUELME(7) FISIOTERAP. ROGUER CAAMAÑO(7) CLUB PANCREAS CHILE DRA CECILIA CASTILLO DR ZOLTAN BERGER DR RODRIGO PONCE
 CENTRO GASTROENTEROLOGICO LOS ARRAYANES (ONG) CLUB DEL PANCREAS: . DRA CECILIA CASTILLO. CLINICA ALEMANA STAGO. . DR ZOLTAN BERGER. CLINICA ALEMANA. . DR RODRIGO PONCE. INSTITUTO CHILENO JAPONES DEL. CANCER . HOSP SAN BORJA ARRIARÁN. DR FERNANDO KAWAGUCHI(1, 2) DR RODRIGO LOAIZA(1,3) DR FERNANDO RIQUELME(1,3) DR CARLOS BRICEÑO(1,2) DR GONZALO ZULOAGA (1,3) DR GERMAN ABRIGÓ(1,3) DR PATRICIO TORRES (1 ) MG JL CASTILLO (4) AL CRISTIAN VASQUEZ PARRA (1) AL MARCOS AVILA (5) AL CYNTHIA PEREZ (1) ING. 3D ALEJANDRO SEPULVEDA(6,7) COORD LILIAN SOTO(6,7) NUTRIC. BLANCA BOERO(7) SICOL. LISETTE RIQUELME(7) FISIOTERAP. ROGUER CAAMAÑO(7) CLUB PANCREAS CHILE. DRA CECILIA CASTILLO. DR ZOLTAN BERGER. DR RODRIGO PONCE.")
2
64 pacientes (promedio 52 años de edad) con pancreatitis aguda
o crónica reagudizada y pacientes con pancreatitis crónica fueron prospectivamente randomizados para recibir BUPRENORFINA más analgésicos no opioides o Placebo más analgésicos no opioides. Placebo y BUPRENORFINA se administraron en infusiones intravenosas continuas en pancreatitis aguda y por vía epidural en pancreatitis crónica METODOS 1 Los analgésicos no opioides se administraron por vía oral según demanda las infusiones intravenosas continuas y por vía epidural de BUPRENORFINA Se evaluó severidad de la pancreatitis según parámetros clínicos PCR y TAC Y sus efectos colaterales que la evidencian como una droga de riesgo Administrada por esta vía. METODOS 1 Se evaluó el grado de dolor según escala análoga visual En pancreatitis aguda se evaluó después del tercer día En pancreatitis crónica se evaluó después de 6 meses RESULTADOS: Aquellos pacientes tratados con BUPRENORFINA TRANSDERMICA LUEGO DE 24 HORAS ( 20 PACIENTES CON P.A. BILIAR O POR HIPERTRIGLICERIDEMIA CON TTO OPIACEO EV Y LUEGO DE LAS 24 HORAS HASTA 2-7 SEMANAS CON 35 ug/hr , desde ¼ a 11/2 parche con cambio a las 72 horas y refuerzo en gotas ocasional con tramal gotas, en las 1eras 72 horas, logró bajar a EVA 0 en 6 de los 18 pacientes (Promedio: EVA 5 a las 48 hrs y luego manteniendo un plateau, bajando a EVA 2-3 a las 72 hrs y logrando su alta antes de las 96 horas , demostrando en este grupo algo más impresionante Cual fue la disminución acelerada de las enzimas pancreáticas y de los marcadores de severidad : 4 de ellos demostraron una disminución en el grado de inflamación pancreática y marcadores de inflamación Esto ocurrió siempre en P.A. Leve a Moderada y con ERCP Terapéutica antes de las 24 horas(p<0.001) No ocurrió lo mismo en pacientes con P.A. Severa y con evolución mayor a las 24 horas(p<0.005) La diferencia en relación al EVA e Inflamación fue significativa en aquellos pacs tratados con placebo y analgésicos no opioides Las dosis de analgésicos no opioides fueron considerablemente menores en los pacientes a a los cuales se les administró BUPRENORFINA. No hubo diferencias significativas entre P.A. Biliar,con ERCP terapeutico de las P.A. post hipertrigliceridemia.
 No ocurrió lo mismo en pacientes con P.A. Severa y con evolución mayor a las 24 horas(p<0.005) La diferencia en relación al EVA e Inflamación fue significativa en aquellos pacs tratados con placebo. y analgésicos no opioides Las dosis de analgésicos no opioides fueron considerablemente menores en. los pacientes a a los cuales se les administró BUPRENORFINA. No hubo diferencias significativas entre P.A. Biliar,con ERCP terapeutico. de las P.A. post hipertrigliceridemia.")
3
Agonista-Antagonista según dosis Receptores MU, KAPPA y DELTA
Vida media muy prolongada. Es un derivado N-ciclopropilmetil de la oripavina Efecto de larga duración: 4-8 horas Metabolismo hepático por glucuronidación Tiene bajo perfil de adicción y dependencia probablemente debido a sus propiedades agonistas parciales y sus características farmacocinéticas de muy lenta disociación desde el receptor mu Determinar si el tratamiento con BUPRENORFINA logra reducir la severidad de la pancreatitis Conocer el efecto analgésico de BUPRENORFINA en pancreatitis aguda y crónica Conocer la importancia de los receptores mu En pancreatitis aguda y crónica Determinar dependencia de dosis en el efecto analgésico HIPOTESIs “Como la activación del cimógeno tiene un rol central en la patogénesis de la pancreatitis aguda, minimizar la cantidad de cimógeno podría reducir la severidad de la PA. La Buprenorfina podría inhibir la síntesis de enzimas en las células acinares, por lo cual reduciría la gravedad de la pancreatitis” La pancreatitis aguda y crónica lleva a aumento de la señalización nociceptiva en T9 y T10 que es determinante en la sensación dolorosa, por lo que la BUPRENORFINA mediante la ctivación directa de receptores opioides centrales podría bloquear la sensación dolorosa A mayor dosis de BUPRENORFINA se lograría mayor bloqueo de receptores mu y así un mayor efecto analgésico/ANTIINFLAMATORIO

4
Conclusiones BUPRENORFINA tiene un efecto moderador en la severidad de la pancreatitis La señalización nociceptiva es bloqueada por BUPRENORFINA mediante la activación directa de receptores mu centrales Corroboramos además estudios realizados por otros autores quienes observaron una menor estadía en la hospitalización De los pacientes con P.A. especialmente en aquellas Crisis Recurrentes

5
Conclusiones El efecto analgésico provocado por BUPRENORFINA
es similar en pancreatitis aguda y crónica El efecto analgésico de la BUPRENORFINA es dosis dependiente

6
Resultados: Todos los pacientes con P. A
Resultados: Todos los pacientes con P.A.Leve aliviaron el dolor considerablemente en el transcurso de los 3 días de tratamiento(p<0.0001) RESULTADOS: Todos los pacientes con pancreatitis crónica, y con lesiones sólido quísticas con crisis recurrentes aliviaron el dolor y la calidad de vida, sólo después de 2-6 meses de tratamiento, con dosis de alrededor de 1- 2 parches cada 72 horas.(p<005) El alivio del dolor fue mayor en pacientes que recibieron mayores dosis Todos comienzan su efecto clínico no antes de las 8-12 horas Siendo mucho más efectivo con dosis mayores a ½ parche de 35 ug/hr Pero , requirió en 1 de 3 pacs, un manejo agresivo del estado nauseoso. Esto no ocurrió si la dosis fue aumentandose progresivamente cada 24 horas NOTA: EL MODELO EXPERIMENTAL EN LAS CÉLULAS DE LOS CONDUCTOS PANCREATICOS, MODIFICÓ NOTABLEMENTE LA SEVERIDAD DE LA INFLAMACIÓN CON EVIDENTE DISMINUCIÓN EN LOS ZIMÓGENOS DE MIGRACIÓN Y TRANSLOCACION INTRACELULAR. ESTO DEMUESTRA SU POTENCIAL UTILIDAD EN LAS UCI CON PACS PORTADORES DE PANCREATITIS SEVERA MÄS ALLÄ DE LA ANALGESIA YA CONOCIDA , Todo lo anterior esta de acuerdo con : Hirata K, (Nippon Rinsho 2004, Review ISSN ; Ogden JM (Dig Dis and Sciences :11 ( ), ISSN ; Gil Cebriana J (Medic Intensiv : 2 ( ),ISSN ;
 RESULTADOS: Todos los pacientes con pancreatitis crónica, y con lesiones sólido quísticas con crisis recurrentes aliviaron el dolor y la calidad de vida, sólo después de 2-6 meses de tratamiento, con dosis de alrededor de 1- 2 parches cada 72 horas.(p<005) El alivio del dolor fue mayor en pacientes que recibieron mayores dosis. Todos comienzan su efecto clínico no antes de las 8-12 horas. Siendo mucho más efectivo con dosis mayores a ½ parche de 35 ug/hr. Pero , requirió en 1 de 3 pacs, un manejo agresivo del estado nauseoso. Esto no ocurrió si la dosis fue aumentandose progresivamente cada 24 horas. NOTA: EL MODELO EXPERIMENTAL EN LAS CÉLULAS DE LOS CONDUCTOS PANCREATICOS, MODIFICÓ NOTABLEMENTE LA SEVERIDAD DE LA INFLAMACIÓN CON EVIDENTE DISMINUCIÓN EN LOS ZIMÓGENOS DE MIGRACIÓN Y TRANSLOCACION INTRACELULAR. ESTO DEMUESTRA SU POTENCIAL UTILIDAD EN LAS UCI CON PACS PORTADORES DE PANCREATITIS SEVERA MÄS ALLÄ DE LA ANALGESIA YA CONOCIDA , Todo lo anterior esta de acuerdo con : Hirata K, (Nippon Rinsho 2004, Review ISSN ; Ogden JM (Dig Dis and Sciences :11 ( ), ISSN ; Gil Cebriana J (Medic Intensiv : 2 ( ),ISSN ;")
7
BUPRENORFINA EN EL TRATAMIENTO DEL DOLOR POR PANCREATITIS AGUDA
TRABAJO DE INVESTIGACION EN 64 PACIENTES PORTADORES DE PANCREATITIS CRONICA Y AGUDA EN HOSPITAL DEL TRABAJADOR DE CONCEPCION SE REALIZÓ ESTE TRABAJO EN RELACION A PACIENTES HOSPITALIZADOS EN EL HOSPITAL DEL TRABAJADOR DE CONCPCION Y DEL HOSPITAL REGIONAL DE CONCEPCION EN EL TRANSCURSO DE ESTOS ULTIMOS 12 MESES TANTO EN PACIENTES CON DOLOR PANCREATICO POR CUADROS ONCOLÓGICOS COMO DOLOR CRÓNICO POR PANCREATITIS CRÓNICA Y DOLOR POR PANCREATITIS AGUDA CON CUADROS DE COLANGITIS Y/O COLEDOCOLITIASIS AGREGADA. SE CORRELACIONÓ EL EFECTO DE ESTE MEDICAMENTO EN RELACIÓN A LA MORFINA Y/O DEMEROL ENDOVENOSO. CINCUENTA Y DOS PACIENTES PORTADORES DE DOLOR CRÓNICO O DOLOR AGUDO RECURRENTE EN PACIENTES CON PATOLOGÍA BENIGNA O MALIGNA FUERON SOMETIDOS A ESTE ESTUDIO BAJO CONSENTIMIENTO INFORMADO. EN RELACIÓN AL DOLOR AGUDO POST PATOLOGÍA BILIAR, SOMETIDOS A TRATAMIENTO CON ESFINTEROTOMÍA Y DRENAJE DE LA VÍA BILIAR(20 DE LOS 64 PACIENTES) ESTE FUE ADMINISTRADO LUEGO DE HORAS POSTERIOR AL TRATAMIENTO CON DEMEROL POR VÍA ENDOVENOSA. (DEBIDO AL TIEMPO QUE DEMORA EN ACTUAR EL PARCHE DE BUPRENORFINA QUE EN NUESTRA EXPERIENCIA VARIÓ ENTRE 8-12 HORAS POSTERIOR A LA APLICACIÓN DE UN PARCHE DE 35 ug/h. ES NECESARIO UN APOYO ANALGÉSICO INICIAL CON OPIÁCEOS POR VÍA EV SE APLICÓ UN CUARTO DE PARCHE A UN PARCHE ENTERO DEPENDIENDO DE LA ESCALA ANÁLOGA DEL DOLOR UTILIZADA (EN ESTE CASO FUE LA ESCALA DE EVA),
 ESTE FUE ADMINISTRADO LUEGO DE HORAS POSTERIOR AL TRATAMIENTO CON DEMEROL POR VÍA ENDOVENOSA. (DEBIDO AL TIEMPO QUE DEMORA EN ACTUAR EL PARCHE DE BUPRENORFINA QUE EN NUESTRA EXPERIENCIA VARIÓ ENTRE 8-12 HORAS POSTERIOR A LA APLICACIÓN DE UN PARCHE DE 35 ug/h. ES NECESARIO UN APOYO ANALGÉSICO INICIAL CON OPIÁCEOS POR VÍA EV. SE APLICÓ UN CUARTO DE PARCHE A UN PARCHE ENTERO DEPENDIENDO DE LA ESCALA ANÁLOGA DEL DOLOR UTILIZADA (EN ESTE CASO FUE LA ESCALA DE EVA),")
8
BUPRENORFINA EN EL TRATAMIENTO DEL DOLOR POR PANCREATITIS AGUDA
RESULTADOS: AQUELLOS PACIENTES QUE RECIBIERON BUPRENORFINA DEMANDARON MENOS ANALGESIA COMPLEMENTARIA EN RELACIÓN A AQUELLOS QUE NO ENTRARON EN EL PROTOCOLO CON LOS PARCHES DE BUPRENORFINA(P< 0.001). LA ESCALA ANÁLOGA DE E.V.A. UTILIZADA PARA EVALUAR LA INTENSIDAD DEL DOLOR EN LOS PRIMEROS SIETE DÍAS, FUE SIGNIFICATIVAMENTE MENOR EN AQUELLOS PACIENTES EN LOS QUE SE UTILIZÓ BUPRENORFINA CUTÁNEA(SE CONTÓ CON LA ASESORÍA DE UNA ENFERMERA DEL DEPARTAMENTO DE ENFERMERÍA (SRA OLIVIA SANHUEZA) PARA CHEQUEAR A CADA PACIENTE EN RELACIÓN A LA INTENSIDAD DEL DOLOR Y SU CALIDAD DE VIDA , ADEMÁS DE LOS EFECTOS COLATERALES. LA REDUCCIÓN DEL DOLOR FUE MÁS INTENSA EN LOS 3 PRIMEROS DÍAS DE UTILIZADO ESTE ANALGÉSICO CUTÁNEO (P<0.001). LOS EFECTOS COLATERALES NO VARIARON SIGNIFICATIVAMENTE EN UNO Y OTRO GRUPO, EXCEPTO POR LAS NAUSEAS EN 4 PACIENTES QUE LOGRÓ CONTROLARSE CON DOMPERIDONA RECTAL EN DOSIS DE 60 MG CADA 8 HORAS. ESTOS EFECTOS SOBRE EL DOLOR, DISMINUYERON SIGNIFICATIVAMENTE LOS DÍAS DE HOSPITALIZACIÓN Y LA CALIDAD DE VIDA INMEDIATAMENTE POSTERIOR, PROLONGÁNDOSE SU USO HASTA 2 -3 SEMANAS POSTERIOR A SU USO COMPLEMENTARIO LAS PRIMERAS HORAS DE INICIADO EL CUADRO AGUDO.
. LA ESCALA ANÁLOGA DE E.V.A. UTILIZADA PARA EVALUAR LA INTENSIDAD DEL DOLOR EN LOS PRIMEROS SIETE DÍAS, FUE SIGNIFICATIVAMENTE MENOR EN AQUELLOS PACIENTES EN LOS QUE SE UTILIZÓ BUPRENORFINA CUTÁNEA(SE CONTÓ CON LA ASESORÍA DE UNA ENFERMERA DEL DEPARTAMENTO DE ENFERMERÍA (SRA OLIVIA SANHUEZA) PARA CHEQUEAR A CADA PACIENTE EN RELACIÓN A LA INTENSIDAD DEL DOLOR Y SU CALIDAD DE VIDA , ADEMÁS DE LOS EFECTOS COLATERALES. LA REDUCCIÓN DEL DOLOR FUE MÁS INTENSA EN LOS 3 PRIMEROS DÍAS DE UTILIZADO ESTE ANALGÉSICO CUTÁNEO (P<0.001). LOS EFECTOS COLATERALES NO VARIARON SIGNIFICATIVAMENTE EN UNO Y OTRO GRUPO, EXCEPTO POR LAS NAUSEAS EN 4 PACIENTES QUE LOGRÓ CONTROLARSE CON DOMPERIDONA RECTAL EN DOSIS DE 60 MG CADA 8 HORAS. ESTOS EFECTOS SOBRE EL DOLOR, DISMINUYERON SIGNIFICATIVAMENTE LOS DÍAS DE HOSPITALIZACIÓN Y LA CALIDAD DE VIDA INMEDIATAMENTE POSTERIOR, PROLONGÁNDOSE SU USO HASTA 2 -3 SEMANAS POSTERIOR A SU USO COMPLEMENTARIO LAS PRIMERAS HORAS DE INICIADO EL CUADRO AGUDO.")
9
BUPRENORFINA EN EL TRATAMIENTO DEL DOLOR POR PANCREATITIS AGUDA
Resumen: • Buprenorfina es un analgésico opióide de bajo peso molecular y lipofílico. Recientemente, se dispone de una fórmulación transdérmica diseñada para liberar buprenorfina a 35, 52.5 y 70 µg/h durante un periodo de 72 horas.• En un ensayo doble ciego, controlado con placebo y aleatorizado, se obtuvo una analgesia al menos satisfactoria con una mínima necesidad de medicación de rescate en el 34%-50% de los pacientes con dolor crónico tratados con buprenorfina transdérmica 35, 52.5 y 70 µg/h y en el 31% de los que recibieron placebo. • En un estudio realizado en pacientes tratados sin éxito con opioides débiles o morfina, el 36.6% y el 47.5% de los tratados con buprenorfina, 35µg/h y 52.5µg/h, respectivamente, experimentaron una analgesia al menos satisfactoria y recibieron buprenorfina sublingual frente al 16.2% de los tratados con placebo (ambos valores de p<0.032). • En dos ensayos, la necesidad de medicación de rescate disminuyó en >50% de los pacientes tratados con buprenorfina transdérmica. Además, a pesar de la disponibilidad de medicación de rescate para todos los pacientes, los que recibieron buprenorfina transdérmica experimentaron una mayor alivio del dolor, una menor intensidad del dolor y un suelo más prolongado sin dolor.
. • En dos ensayos, la necesidad de medicación de rescate disminuyó en >50% de los pacientes tratados con buprenorfina transdérmica. Además, a pesar de la disponibilidad de medicación de rescate para todos los pacientes, los que recibieron buprenorfina transdérmica experimentaron una mayor alivio del dolor, una menor intensidad del dolor y un suelo más prolongado sin dolor.")
11
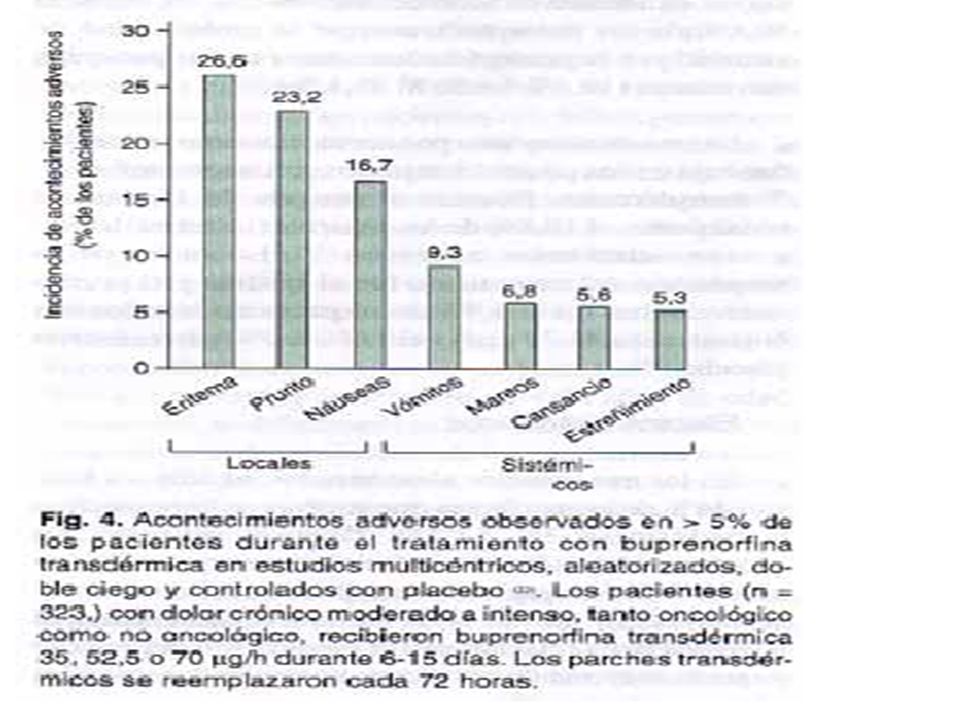
• En general, la buprenorfina transdérmica fue bien tolerada
• En general, la buprenorfina transdérmica fue bien tolerada. Los acontecimientos adversos sistémicos fueron los típicos asociados al tratamiento con opioides o se atribuyeron a la Enfermedad subyacente. En los últimos años, se han publicado directrices más detalladas sobre el abordaje del dolor crónico (l 4), aunque en muchos pacientes con este tipo de dolor el tratamiento es incorrecto y el alivio, insuficientes (5). Las directrices de la OMS sobre el tratamiento del dolor oncológico aconsejan el uso de una escalera farmacológica de tres peldaños basada en la intensidad del dolor (6). En caso de dolor leve, se recomienda un analgésico no opioide, para el dolor de intensidad de leve a moderada puede añadirse un opioide débil, como tramadol, mientras que para el dolor moderado o intenso, se utilizarán opioides fuertes, como morfina, metadona, fentanilo y buprenorfina(5). Más recientemente, se han empezado a usar opioides en el tratamiento del dolor crónico no oncológico (7-9).
, aunque en muchos pacientes con este tipo de dolor el tratamiento es incorrecto y el alivio, insuficientes (5). Las directrices de la OMS sobre el tratamiento del dolor oncológico aconsejan el uso de una escalera farmacológica de tres peldaños basada en la intensidad del dolor (6). En caso de dolor leve, se recomienda un analgésico no opioide, para el dolor de intensidad de leve a moderada puede añadirse un opioide débil, como tramadol, mientras que para el dolor moderado o intenso, se utilizarán opioides fuertes, como morfina, metadona, fentanilo y buprenorfina(5). Más recientemente, se han empezado a usar opioides en el tratamiento del dolor crónico no oncológico (7-9).")
12
Las alternativas terapéuticas
Para el dolor crónico han mejoadogracias al desarrollo de nuevas formasde administración (10). Una de éstas es el sistema de administración transdérmica de fármacos que ofrece varias ventajas sobre las vías parenteral y oral (11-12). En primer lugar, un sistema de administración transdérmica evita la molestia de inyecciones intramusculares múltiples o el rechazo del paciente a tomar preparados orales y no requiere personal sanitario como en el caso de las infusiones intravenosas(11). En segundo lugar, la liberación transdérmica del fármaco produce concentraciones plasmáticas del fármaco estables debido a su liberación controlada y evita los inconvenientes derivados del metabolismo hepático de primer paso, la pobre absorción en el tracto gastrointestinal y la existencia deuna biodisponibilidad baja o variable según el paciente (11, 12). Además, los sistemas de liberación transdérmica reducen la frecuencia de administración (lo que podría mejorar el cumplimiento terapéutico) y permiten cambiar regularmente el sitio de aplicación, con la consiguiente disminución del riesgo de reacciones adversas locales (12, 13).
. Una de éstas es el sistema de administración transdérmica de fármacos que ofrece varias ventajas sobre las vías parenteral y oral (11-12). En primer lugar, un sistema de administración transdérmica evita la molestia de inyecciones intramusculares múltiples o el rechazo del paciente a tomar preparados orales y no requiere personal sanitario como en el caso de las infusiones intravenosas(11). En segundo lugar, la liberación transdérmica del fármaco produce. concentraciones plasmáticas del fármaco estables debido a su liberación controlada y evita los inconvenientes derivados del metabolismo hepático de primer paso, la pobre absorción en el tracto gastrointestinal y la existencia deuna biodisponibilidad baja o variable según el paciente (11, 12). Además, los sistemas de liberación transdérmica reducen la frecuencia de administración (lo que podría mejorar el cumplimiento terapéutico) y permiten cambiar regularmente el sitio de aplicación, con la consiguiente disminución del riesgo de reacciones adversas locales (12, 13).")
17
• Al menos un tercio de los pacientes tratados con buprenorfina transdérmica respondieron al tratamiento en dos ensayos aleatorizados (figura 2) (37, 38). Cabe destacar que > 33% de los tratados sin éxito con opioides débiles o morfina respondieron al tratamiento con buprenorfina transdérmica y que el porcentaje de pacientes con respuesta en los grupos de tratamiento con 35 y 52,5 gg/h fue significativamente mayor que en los que recibieron placebo en este estudio (38).
.")
18
REDUCCION ANALGESIA BASICA
La administración adicional de analgésicos opioides orales se redujo en >50% con buprenorfina transdérmica. Para los grupos de tratamiento activo todas la reducciones en la necesidad de medicación de rescate fueron significativamente mayores (p< 0,05) que las observadas en los que recibieron placebo (8%; ) (38). • La necesidad de medicación de rescate también se relacionó con la necesidad de buprenorfina sublingual en el periodo basal (39). En el estudio que comparó buprenorfina transdérmica 35 µg/h con placebo se observó que en pacientes que recibían 0,4-0,8, 0,8-1,2, 1,2-1,6 ó y 1,6 mg de buprenorfina sublingual durante el periodo de preinclusión, las necesidades de medicación de rescate durante el periodo de tratamiento de buprenorfina transdérmica fue de 0,2, 0,4, 0,6 y 0,9 mg, respectivamente (39). Cuando se combinó la cantidad de buprenorfina utilizada como medicación de rescate con la dosis diaria administrada mediante el parche de buprenorfina transdérmica (0,8 mg), la dosis total de buprenorfma administrada fue casi exactamente igual a la dosis recibida durante el periodo de preinclusion (42).
 que las observadas en los que recibieron placebo (8%; ) (38). • La necesidad de medicación de rescate también se relacionó con la necesidad de buprenorfina sublingual en el periodo basal (39). En el estudio que comparó buprenorfina transdérmica 35 µg/h con placebo se observó que en pacientes que recibían 0,4-0,8, 0,8-1,2, 1,2-1,6 ó y 1,6 mg de buprenorfina sublingual durante el periodo de preinclusión, las necesidades de medicación de rescate durante el periodo de tratamiento de buprenorfina transdérmica fue de 0,2, 0,4, 0,6 y 0,9 mg, respectivamente (39). Cuando se combinó la cantidad de buprenorfina utilizada como medicación de rescate con la dosis diaria administrada mediante el parche de buprenorfina transdérmica (0,8 mg), la dosis total de buprenorfma administrada fue casi exactamente igual a la dosis recibida durante el periodo de preinclusion (42).")
19
A pesar de la disponibilidad de medicación de rescate para todos los pacientes, los que recibieron buprenorfina transdérmica tendieron a experimentar un mayor alivio del dolor que los tratados con placebo (37-39). En uno de los ensayos, el porcentaje de pacientes que manifestaron un alivio del dolor de bueno a total aumentó hasta en un 13,2% y 8,6% durante la aplicación del primer y segundo parche de buprenorfina transdérmica, respectivamente, frente a una reducción del 20% y el 11,4% en los pacientes que recibieron placebo (37). Durante los 15 días de tratamiento, el 40,4%-46,3% de los pacientes tratados con Buprenorfina transdérmica tuvieron un alivio del dolor entre bueno y total en comparación con el 32,4% de los que recibieron placebo(38). Esto también se reflejó en la media de las puntuaciones del alivio del dolor registradas por los pacientes durante el estudio; las medias de las puntuaciones verbales de evaluación (determinadas mediante una escala de cuatro puntos [1-4: escaso, satisfactorio, bueno o total]) fueron de 2,3, 2,4 y 2,5 en los pacientes tratados con buprenorfma transdérmica 35, 52,5 y 70 µg/h, respectivamente, frente a 1,9 en los que recibieron placebo (38). •
. Durante los 15 días de tratamiento, el 40,4%-46,3% de los pacientes tratados con Buprenorfina transdérmica tuvieron un alivio del dolor entre bueno y total en. comparación con el 32,4% de los que recibieron placebo(38). Esto también se reflejó en la media de las puntuaciones del alivio del dolor registradas por los pacientes durante el estudio; las medias de las puntuaciones verbales de evaluación (determinadas mediante una escala de cuatro puntos [1-4: escaso, satisfactorio, bueno o total]) fueron de 2,3, 2,4 y 2,5 en los pacientes tratados con buprenorfma transdérmica 35, 52,5 y 70 µg/h, respectivamente, frente a 1,9 en los que recibieron placebo (38). •")
20
Oncology (Huntingt) 2000 Nov; 14 (11 A):135-50
BIBLIOGRAFIA 1. Ferrelí B, Casarett D, Epplin J, et al. The management of persistent pain in older persons. J Am Geriatr Soc 2002; 50 (6 Suppl.): S205-24 2. Lawhome L, Passerini J, Cranmer K, et al.Chronic pain management in the long-term care setting. Columbia (MD): American Medical Directors Association, 1999 3. Benedetti C, Brock C, Cleeland C, et al. NCCN Practice Guidelines for Cancer Pain. Oncology (Huntingt) 2000 Nov; 14 (11 A):135-50 4. Jacobi J, Fraser GL, Coursin DB, et al.Clinical practice guidelmes for the sustained use of sedatives and analgesics in the critically ilí adult. Crit Care Med 2002;30: 5. Control of cancer pain often madequate despite effective drugs and guidelines. Drug Ther Perspect 2002 Feb; 18(2): World Health Organisation. WHO’s pain relief ladder [oníme]. Available from URL: May 15] 7. Graziotti PJ, Goucke CR. The use of oral opioids in patients wih chronic non-cancer pain: management strategies. Med J Aust 1997; 167: 30-4 8. Simpson KH. Individual choice of opioids and formulations: Strategies to achieve the optimum for the patient. Clin heumatol 2002; 21 (1 Suppl.): S5-8 9. Bannwarth B. Risk-benefit assessment of opioids m chronic noncancer pain. Drug Saf 1999; 21(4): 10. Grond S, Radbruch L, Lehmann KA.Clinical pharmacokinetics of transdermal opioids. focus on transdermal fentanyl.Clin Pharmacokinet 2000; 38 (1): 59-89 11. Caplan RA, Southam M. Transdermal drug delivery and its application to pain control. In: Benedetti C, editor. Advances in pain research and therapy. New York: Rayen Press, 1990; 14: 12. Benson HAE, Prankerd RJ. Optimisation of drug delivery 4: transdermal drug delivery. Aust J Hosp Pharm 1997; 27 (6): 441-8
: S Lawhome L, Passerini J, Cranmer K, et al.Chronic pain management in the long-term. care setting. Columbia (MD): American Medical Directors Association, Benedetti C, Brock C, Cleeland C, et al. NCCN Practice Guidelines for Cancer Pain. Oncology (Huntingt) 2000 Nov; 14 (11 A): Jacobi J, Fraser GL, Coursin DB, et al.Clinical practice guidelmes for the sustained use of sedatives and analgesics in the critically ilí adult. Crit Care Med 2002;30: Control of cancer pain often madequate despite effective drugs and guidelines. Drug Ther Perspect 2002 Feb; 18(2): World Health Organisation. WHO’s pain relief ladder [oníme]. Available from URL: May 15] 7. Graziotti PJ, Goucke CR. The use of oral opioids in patients wih chronic non-cancer pain: management strategies. Med J Aust 1997; 167: Simpson KH. Individual choice of opioids and formulations: Strategies to achieve the optimum for the patient. Clin heumatol. 2002; 21 (1 Suppl.): S Bannwarth B. Risk-benefit assessment of opioids m chronic noncancer pain. Drug Saf 1999; 21(4): Grond S, Radbruch L, Lehmann KA.Clinical pharmacokinetics of transdermal opioids. focus on transdermal fentanyl.Clin Pharmacokinet 2000; 38 (1): Caplan RA, Southam M. Transdermal drug delivery and its application to pain control. In: Benedetti C, editor. Advances in pain research and therapy. New York: Rayen. Press, 1990; 14: Benson HAE, Prankerd RJ. Optimisation of drug delivery 4: transdermal drug delivery. Aust J Hosp Pharm 1997; 27 (6):")
21
13. Berner B, John VA. Pharmacokinetic characterisation of transdermal delivery systems. Clin Pharmacokinet 1994; 26 (2):121-34 14. Ripamonti C, Dickerson ED. Strategies for the treatment of cancer pain in the new miliennium. Drugs 2001; 61(7): 15. Roth SH. A new role for opioids in the treatment of arthritis. Drugs 2002; 62 (No. 2): 16. ABPI Medicines Compendium. Surrey:Datapharm Communications Ltd, 2003 17. Heel RC, Brogden RN, Speight TM, et al. Buprenorphine. A review of its pharmacological properties and therapeutic efficacy. Drugs 1979; 17: 18. Stem M. New transdermal pam therapy with buprenorphine [m German]. Dtsch Apoth Ztg 2000; 140 (27): 31 19. Radbruch L. A therapeutics masterclass. Eur J Palliat Care 2003; 10 (1 Suppl.): Grunenthal GmbH. Transtec (R) scientific monograph. Aachen: Grunenthal GmbH, 2002 21. American Society of Health-System Pharmacists. AHFS Drug Information.2003 22. Jasinski DR, Pevnick JS, Griffith JD. Human pharmacology and abuse potencial of the analgesia buprenorphine: a potencial agent for treating narcotic addiction. Arch Gen Psychiatry 1978; 35 (4): 23. Sittl R. Buprenorphine transdermal patch: clinical expert report. Germany: Grunenthal GmbH, 2000
: Roth SH. A new role for opioids in the treatment of arthritis. Drugs 2002; 62 (No. 2): ABPI Medicines Compendium. Surrey:Datapharm Communications Ltd, Heel RC, Brogden RN, Speight TM, et al. Buprenorphine. A review of its pharmacological properties and therapeutic efficacy. Drugs 1979; 17: Stem M. New transdermal pam therapy with buprenorphine [m German]. Dtsch Apoth Ztg 2000; 140 (27): Radbruch L. A therapeutics masterclass. Eur J Palliat Care 2003; 10 (1 Suppl.): Grunenthal GmbH. Transtec (R) scientific monograph. Aachen: Grunenthal GmbH, American Society of Health-System Pharmacists. AHFS Drug Information Jasinski DR, Pevnick JS, Griffith JD. Human pharmacology and abuse potencial. of the analgesia buprenorphine: a potencial agent for treating narcotic addiction. Arch. Gen Psychiatry 1978; 35 (4): Sittl R. Buprenorphine transdermal patch: clinical expert report. Germany: Grunenthal. GmbH,")
22
24. Cowan A, Lewis 1W, Macfarlane IR
24. Cowan A, Lewis 1W, Macfarlane IR.Agonist and antagonist properties of buprenorphine, a new antinociceptive agent. Br 3 Pharmacoí 1977; 60 (4):537-45 25. Budd K. Evidence based medicine in practice. Buprenorphme: a review.Newmarket: hayward Medical Communications, 2002 26. Walsh SL, Preston KL. Stitzer ML, et al.Clinical pharmacology of buprenorphine: Ceiling effects at high doses. Clin Phannacol Ther 1994; 55: 27. Zaki PA, Keith DE, Brine GA, et al. Ligand-induced changes in surface l.topioid receptor number: relationship to G protein activation? J Pharmacol Exp Ther (3): 28. Orwm JM. Pharmacological aspects in man. In: Harcus AW, Smith RB, Whittte BA, editors. PAINNew Perspectives in Measurement and Management. Edinburgh: Churchill Livingstone, 1977: 29. Downing 1W, Goodwin NM, Hicks 3. The respiratory depressive effects of intravenous buprenorphine in patients in an intensive care unit. 5 Afr Med J 1979; 55 (25): 30. Budd K. High dose buprenorphine for postoperative analgesia. Anaesthesia 1981; 36: 900-3 31. Walsh SL, Preston KL, Bigelow GE, et al. Acute administration of buprenorphine in humans: Partial agonist and blockade effects. J Pharmacol Exp Ther 1995; 274 (1): 32. Terlinden R. Pupillometry data aspharmacodynamic paramter from a pharmacokinetic study on single application of buprenorphine transdermal system TDS) [data on file]. Aachen: Grunenthal GmbH,2000 33. Terlinden R, Stadler T. Pharmacokinetic study on single application of buprenorphme transdermal system (TDS) [data on file]. Aachen: Grunenthal GmbH,2000 34. Budd K. Buprenorphine and the transdermal system: the ideal match in pain management. Int 3 Clin Pract Suppl 2003;(1 33 Suppl,): 9-14 35. Data on file. Grünenthal, Masche UP. Transdermal buprenorphine [in German]. Pharma Kritik 2001; 23 (11): 43-4
: Budd K. Evidence based medicine in practice. Buprenorphme: a review.Newmarket: hayward Medical Communications, Walsh SL, Preston KL. Stitzer ML, et al.Clinical pharmacology of buprenorphine: Ceiling effects at high doses. Clin Phannacol Ther 1994; 55: Zaki PA, Keith DE, Brine GA, et al. Ligand-induced changes in surface l.topioid receptor number: relationship to G protein activation J Pharmacol Exp Ther (3): Orwm JM. Pharmacological aspects in man. In: Harcus AW, Smith RB, Whittte BA, editors. PAINNew Perspectives in Measurement and Management. Edinburgh: Churchill Livingstone, 1977: Downing 1W, Goodwin NM, Hicks 3. The respiratory depressive effects of intravenous buprenorphine in patients in an intensive care unit. 5 Afr Med J 1979; 55 (25): Budd K. High dose buprenorphine for postoperative analgesia. Anaesthesia 1981; 36: Walsh SL, Preston KL, Bigelow GE, et al. Acute administration of buprenorphine in. humans: Partial agonist and blockade effects. J Pharmacol Exp Ther 1995; 274. (1): Terlinden R. Pupillometry data aspharmacodynamic paramter from a pharmacokinetic study on single application of buprenorphine transdermal system TDS) [data on file]. Aachen: Grunenthal GmbH, Terlinden R, Stadler T. Pharmacokinetic study on single application of buprenorphme transdermal system (TDS) [data on file]. Aachen: Grunenthal GmbH, Budd K. Buprenorphine and the transdermal system: the ideal match in pain management. Int 3 Clin Pract Suppl 2003;(1 33 Suppl,): Data on file. Grünenthal, Masche UP. Transdermal buprenorphine [in German]. Pharma Kritik 2001; 23 (11):")
23
37. Bóhme K, Likar R. Efficacy and tolerability of a new opiod analgesic Formulation, buprenorphine transdermal therapeutie system (TDS), in the treatment of patients with chronic pain: a randomised, doubleblind,placebo-controlled study. Pain Clinic (2): 38. Sittl R, Griessmger N, Likar R. Analgesic efficacy and tolerability of transdermal buprenorphine in patients wíth inadequately controlled chronic pain related to cancer and other disorders: A multicenter, randomized, doubie-blind, placebo-controlled trial. Clin Ther 2003; 25 (1): 39. Sorge 3, Anderson-Hillemacher A.Buprenorphine transdermal system(delivery rate 35 gg/h) in comparison to sublingual tablets in chronic pain patients[abstract plus poster]. 1 st Congress of the Research Network of the European Association for Palliative Care; 2000 Dec 7-9; Berlin 40. Kopp M, Repas C. Buprenorphine transdermal system (TDS) (delivery rate 35J1g/h) in an open long-term study with chronic pain patients [abstract plus poster]. 1 st Congress of the Research Network of the European Association for Palliative Care; 2000 Dec 7-9; Berlin 41. Vielvoye-Kerkmeer APE. Long-term treatment with buprenorphine TDS in patients with chronic pain. Eur J Palliat Care 2003; 10 (1 Suppl.): 17-9 42. Bohme K. Buprenorphine in a rransdermal therapeutic system- a new option. Clin Rheumatol 2002; 21 (1 Suppl.): S13-6 43. Radbruch L, Vielvoye-Kerkmeer A. Buprenorphine TDS: the clinical development- rationale and results. Int J Clin Pract Suppl 2003; (133 Suppl.): 15-8 44. Radbruch L. Efficacy and toíerability of buprcnorphine TDS m cancer pain patients. Eur 3 Palliat Care 2003; 10 (1 Suppl.): Bohme K, Bonjean J. Buprenorphine transdermal system (TTS) (delivery rates 35/52.5/70 gg/h) in comparison with sublingual buprenorphine in chronic pain patients. 1 st Congress of the Research Network of the European Association for Palliative Care; 2000 Dec 7-9; Berlin 46. Anonymous. Buprenorphine transdermal patches launched. Pharm ; 268 7197): 638
: Sittl R, Griessmger N, Likar R. Analgesic efficacy and tolerability of transdermal. buprenorphine in patients wíth inadequately controlled chronic pain related to cancer. and other disorders: A multicenter, randomized, doubie-blind, placebo-controlled trial. Clin Ther 2003; 25 (1): Sorge 3, Anderson-Hillemacher A.Buprenorphine transdermal system(delivery rate 35 gg/h) in comparison to sublingual tablets in chronic pain patients[abstract plus poster]. 1 st Congress of the Research Network of the European Association for Palliative Care; 2000 Dec 7-9; Berlin. 40. Kopp M, Repas C. Buprenorphine transdermal system (TDS) (delivery rate 35J1g/h) in an open long-term study with chronic pain patients [abstract plus poster]. 1 st Congress of the Research Network of the European Association for Palliative. Care; 2000 Dec 7-9; Berlin. 41. Vielvoye-Kerkmeer APE. Long-term treatment with buprenorphine TDS in patients with chronic pain. Eur J Palliat Care 2003; 10 (1 Suppl.): Bohme K. Buprenorphine in a rransdermal therapeutic system- a new option. Clin Rheumatol 2002; 21 (1 Suppl.): S Radbruch L, Vielvoye-Kerkmeer A. Buprenorphine TDS: the clinical development- rationale and results. Int J Clin Pract Suppl 2003; (133 Suppl.): Radbruch L. Efficacy and toíerability of buprcnorphine TDS m cancer pain patients. Eur 3 Palliat Care 2003; 10 (1 Suppl.): Bohme K, Bonjean J. Buprenorphine. transdermal system (TTS) (delivery rates 35/52.5/70 gg/h) in comparison with. sublingual buprenorphine in chronic pain patients. 1 st Congress of the Research. Network of the European Association for Palliative Care; 2000 Dec 7-9; Berlin. 46. Anonymous. Buprenorphine transdermal patches launched. Pharm ; ): 638.")
24
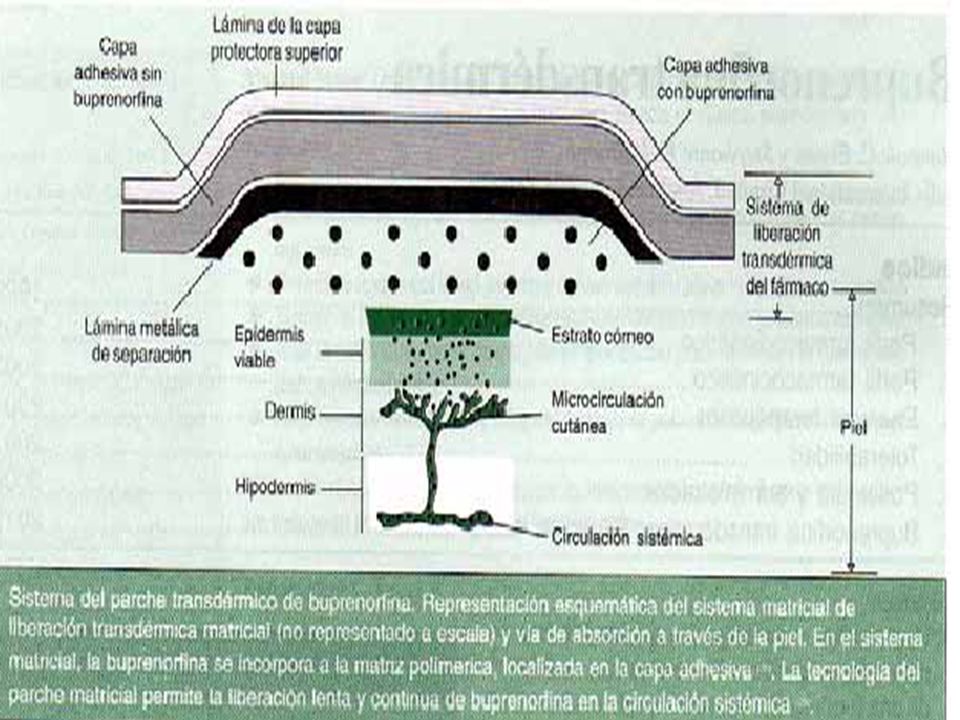
Los analgésicos de liberación lenta cumplen con las recomendaciones para el alivio del dolor (14, 15). Las publicaciones indican que una de las razones del escaso alivio del dolor obtenido en pacientes con cáncer es la prescripción de analgésicos «a demanda» en vez de un tratamiento «pautado o reloj en mano» (14). Además, la OMS recomienda un control «continuo» del dolor crónico no oncológico (15). El parche transdérmico evita este inconveniente al conseguir concentraciones plasmáticas constantes eficaces para obtener una analgesia. Buprenorfina está disponible en una formulación parenteral y sublingual (16) y es un analgésico eficaz en el tratamiento del dolor agudo y crónico (revisión previa en Drugs (17)). Buprenorfína está indicada en el tratamiento del dolor de moderado a intenso y se clasifica como analgésico de tercer escalón en la escalera analgésica de la OMS (5, 18), aunque se ha propuesto como analgésico intermedio entre el escalón 2 y e1 3 (19). Recientemente, se ha desarrollado una formulación de buprenorfina en parches de matriz transdérmica El sistema transdérmico de buprenorfina (TDS; a partir de aquí Denominado buprenorfina transdérmica) se presenta en tres concentraciones: los parches contienen 20, 30 ó 40 mg de buprenorfina y están diseñados para este fármaco a una velocidad controlada de 35, 52.5 y 70 µg/h, respectivamente, cada una correspondiente a la dosis diaria de 0,8, 1,2 y 1,6 mg (20) Todos estos parches se han diseñado para un periodo de aplicación de 72 horas (20), En este artículo se presenta una revisión de los datos clínicos más relevantes sobre la formulación transdérmica de buprenorfina en el tratamiento del dolor crónico.
. Además, la OMS recomienda un control «continuo» del dolor crónico no oncológico (15). El parche transdérmico evita este inconveniente al conseguir concentraciones plasmáticas constantes eficaces para obtener una analgesia. Buprenorfina está disponible en una formulación parenteral y sublingual (16) y es un analgésico eficaz en el tratamiento del dolor agudo y crónico (revisión previa en Drugs (17)). Buprenorfína está indicada en el tratamiento del dolor de moderado a intenso y se clasifica como analgésico de tercer escalón en la escalera analgésica de la OMS (5, 18), aunque se ha propuesto como analgésico intermedio entre el escalón 2 y e1 3 (19). Recientemente, se ha desarrollado una formulación de buprenorfina en parches de matriz transdérmica. El sistema transdérmico de buprenorfina (TDS; a partir de aquí Denominado buprenorfina transdérmica) se presenta en tres concentraciones: los parches contienen 20, 30 ó 40 mg de buprenorfina y están diseñados para este fármaco a una velocidad controlada de 35, 52.5 y 70 µg/h, respectivamente, cada una correspondiente a la dosis diaria de 0,8, 1,2 y 1,6 mg (20) Todos estos parches se han diseñado para un periodo de aplicación de 72 horas (20), En este artículo se presenta una revisión de los datos clínicos más relevantes sobre la formulación transdérmica de buprenorfina en el tratamiento del dolor. crónico.")
25
Perfil farmacodinámico
Buprenorfina es un opioide sintético, lipofilico soluble en agua y de bajo peso molecular; estas propiedades permiten su penetración en los tejidos y su administración transdérmica. Las características farmacodinámicas debuprenorfina, administrada en diversas formulaciones, están bien documentadas y ya se han revisado en Drugs (17), Aquí se presenta un resumen de estas propiedades, además de algunos datos de estudios realizados con el parchetransdérmico. Propiedades de unión a receptores• Buprenorfina es un agonista parcial de los receptores opioides µ y un antagonista de los receptores k en el SNC y en los tejidos periféricos, y posee una elevada afinidad por ambos receptores (21-23). Los efectos analgésicos parecen deberse a su actividad agonista µ(24). Tanto su unión al receptor µ como su disociación de él son lentas, por lo que los efectos de buprenorfina son de comienzo paulatino y de larga duración (21-23). El tiempo hasta el comienzo de la acción y la duración de la actividad de buprenorfina también dependen de la vía de administración (véase la sección 2).
, Aquí se presenta un resumen de estas propiedades, además de algunos datos de estudios realizados con el parchetransdérmico. Propiedades de unión a receptores• Buprenorfina es un agonista parcial de los receptores opioides µ y un antagonista de los receptores k en el SNC y en los tejidos periféricos, y posee una elevada afinidad por ambos receptores (21-23). Los efectos analgésicos parecen deberse a su actividad agonista µ(24). Tanto su unión al receptor µ como su disociación de él son lentas, por lo que los efectos de buprenorfina son de comienzo paulatino y de larga duración (21-23). El tiempo hasta el comienzo de la acción y la duración de la actividad de buprenorfina. también dependen de la vía de administración (véase la sección 2).")
26
• En estudios, la unión de buprenorfina a receptores opioides tiende a seguir una curva dosisrespuesta campaniforme: se observaron incrementos de la eficacia, en función de la dosis, en el rango posológico «más bajo», mientras que con las dosis más altas el efecto no sólo no fue mayor, sino que incluso fue más reducido (revisado por Budd (25)), Aunque no siempre se ha observado una respuesta campaniforme en estudios con voluntarios y pacientes, dicha respuesta si puede ocurrir con dosis superiores a las clínicamente relevantes para la analgesia (25). Por ejemplo, en voluntarios que consumían opioides, pero que no sufrían dependencia física, no se observó un efecto «techo» para la analgesia con la administración de buprenorfina sublingual, de 1-32 mg en dosis ascendentes, dentro del rango de dosis usado para la analgesia (26). Efectos sobre el SNC B• Buprenorfina produce una analgesia que depende de la dosis y es aproximadamente veces más potente que una dosis equivalente (en peso) de morfina (22). Numerosos estudios llevados a cabo en pacientes con dolor oncológico o ostoperatorio han demostrado que la concentración plasmática de buprenorfina requerida para el alivio del dolor moderado a intenso oscila entre 100 y 500 pg/ml (revisado por Sittl (23)).
. Por ejemplo, en voluntarios que consumían opioides, pero que no sufrían dependencia física, no se observó un efecto «techo» para la analgesia con la administración de buprenorfina sublingual, de 1-32 mg en dosis ascendentes, dentro del rango de dosis usado para la analgesia (26). Efectos sobre el SNC B• Buprenorfina produce una analgesia que depende de la dosis y es aproximadamente veces más potente que una dosis equivalente (en peso) de morfina (22). Numerosos estudios llevados a cabo en pacientes con dolor oncológico o ostoperatorio han demostrado que la concentración plasmática de buprenorfina requerida para el alivio del dolor moderado a intenso oscila entre 100 y 500 pg/ml (revisado por Sittl (23)).")
27
• Tras la suspensión del tratamiento con buprenorfina, pueden observarse síntomas de abstinencia (21). Estos síntomas pueden alcanzar su máximo aproximadamente a las 2 semanas, pero parecen ser más leves que los correspondientes al tratamiento con morfina (revisado en Drugs (17)) Además, la probabilidad de desarrollo de dependencia o tolerancia al fármaco después de un tratamiento a corto o largo plazo con buprenorfma puede ser menor que la observada con otros opioides. En un estudio in vitro efectuado en células 293-SF-MOR, se observó que 10 µmol/l de fentanilo y 10 µmol/l de morfina producían una reducción del 35% y el 9%, respectivamente, en los receptores µ de la superficie celular, mientras que 10 µg/l de buprenorfina ocasionaron un incremento del 10% (p <0,05 frente al control) (27). Estos resultados parecen indicar que buprenorfina no induce la intemalización de receptores opioides (pérdida de receptores de la superficie de las células), reduciendo así la probabilidad de desarrollo de tolerancia(27).
) Además, la probabilidad de desarrollo de dependencia o tolerancia al fármaco después de un tratamiento a corto o largo plazo con buprenorfma puede ser menor que la observada con otros opioides. En un estudio in vitro efectuado en células 293-SF-MOR, se observó que 10 µmol/l de fentanilo y 10 µmol/l de morfina producían una reducción del 35% y el 9%, respectivamente, en los receptores µ de la superficie celular, mientras que 10 µg/l de buprenorfina ocasionaron un incremento del 10% (p <0,05 frente al control) (27). Estos resultados parecen indicar que buprenorfina no induce la intemalización de receptores opioides (pérdida de receptores de la superficie de las células), reduciendo así la probabilidad de desarrollo de tolerancia(27).")
28
Resulta interesante destacar la acción sobre receptores muc, y lo poco que se sabe sobre la acción fos-like (número de núcleos inmunoactivados), los niveles de amilasa sérica actividad de la mieloperoxidasa y cerulina plasmática, inhibiendo directamente la acción inflamatoria(Journal Research :2( ). EFECTO DE LA BUPRENORFINA SOBRE LA SINTESIS DE ENZIMAS PANCREATICAS La secreción de enzimas pancreáticas estaría inhibida durante la pancreatitis aguda, resultando en un aumento de los zimógenos intracelulares. Debido a que la activación temprana de los zimógenos ha sido destacada siempre como el punto central de la pancreatitis aguda. La posibilidad de disminuir la cantidad de zimógenos en reservorio acinar, podría logrtar disminuir la respuesta inflamatoria. En relación a los opiáceos , estos tienen una repercusión sobre la secreción que variará dependiendo de la dosis, sitio de administración y la presencia de estimulantes pancreaticos(DIG DIS AND SCIENCES :11( ) El efecto de los opiáceos sobre la pancreatitis aguda es aún desconocida. Estudios experimentales en ratas en las que se inyecta cerulina más buprenorfina y otro grupo sin buprenorfina ha demostrado al medir la masa pancreática incluida el contenido de ADN, amilasa , tripsinógeno, lipasa y quimotripsinógeno, han demostrado recientemente que la buprenorfina reduce el edema pancreático. El contenido de amilasa sérica fue estadísticamente menor debido a la reducción en la síntesis enzimática y por ende en la cantidad de zimógenos acinares pancreáticos.
 El efecto de los opiáceos sobre la pancreatitis aguda es aún desconocida. Estudios experimentales en ratas en las que se inyecta cerulina más buprenorfina y otro grupo sin buprenorfina ha demostrado al medir la masa pancreática incluida el contenido de ADN, amilasa , tripsinógeno, lipasa y quimotripsinógeno, han demostrado recientemente que la buprenorfina reduce el edema pancreático. El contenido de amilasa sérica fue estadísticamente menor debido a la reducción en la síntesis enzimática y por ende en la cantidad de zimógenos acinares pancreáticos.")
29
Otros efectos• No se han etudiado los efectos cardiovasculares de buprenorfina transdérmica; sin embargo, la administración intramuscular, oral o sublingual de buprenorfina disminuye la frecuencia cardíaca y la presión arterialen voluntarios (17-21). Las reducciones en la frecuencia cardiaca tras la administración intramuscular de buprenorfina (0,15-0,6 mg) fueron similares a las observadas con morfina intramuscular (5-12,5 mg) (28). Las variaciones de la frecuencia cardíaca (aunque no de la presión arterial) tras la administración de buprenorfina oral (1- mg) guardaron relación con la dosis. Buprenorfma sublingual (0,4 ó 0,8 mg) produjo descensos de la frecuencia cardíaca junto con un incremento compensatorio del volumen de eyección, por lo que la presión aterial media apenas varió (revisado en Drugs(17))• Con buprenorfina, la depresión respiratoria es infrecuente y raramente tiene trascendencia clínica (21), no obstante el fármaco está contraindiado en pacientes con insuficiencia respiratoria grave (véase la sección 5). En diez pacientes gravemente enfermos, buprenorfma intravenosa (0,2 ó 0,4 mg) redujo la frecuencia respiratoria media e incremento los niveles de dióxido de carbono en sangre arterial, pero no tuvo efectos significativos sobre la frecuencia cardiaca, los niveles de oxígeno en sangre arterial o los valores del exceso de base (29).
. Las reducciones en la frecuencia cardiaca tras la administración intramuscular de buprenorfina (0,15-0,6 mg) fueron. similares a las observadas con morfina intramuscular (5-12,5 mg) (28). Las variaciones de la frecuencia cardíaca (aunque no de la presión arterial) tras la administración de buprenorfina oral (1- mg) guardaron relación con la dosis. Buprenorfma sublingual (0,4 ó 0,8 mg) produjo descensos de la frecuencia cardíaca junto con un incremento compensatorio del volumen de. eyección, por lo que la presión aterial media apenas varió (revisado en Drugs(17))• Con buprenorfina, la depresión respiratoria es infrecuente y raramente tiene trascendencia clínica (21), no. obstante el fármaco está contraindiado en pacientes con insuficiencia respiratoria grave (véase la sección 5). En diez pacientes gravemente enfermos, buprenorfma intravenosa (0,2 ó 0,4 mg) redujo la frecuencia respiratoria media e incremento los niveles de dióxido de carbono en sangre arterial, pero no tuvo efectos significativos sobre la frecuencia cardiaca, los niveles de. oxígeno en sangre arterial o los valores del exceso de base (29).")
30
En voluntarios, la depresión respiratoria con buprenorfina intramuscular fue proporcional a la dosis dentro del rango de 0,5 a 1,2 mg, pero no se consideróclínicamente significativa (28). •. En un estudio de aplicación única efectuado en voluntarios, se observaron reducciones dosisdependientes en el diámetro pupilar tras la aplicación de los parches de 35, 52,5 ó 70 µg/l; la isminución del diámetro de la pupila se mantuvo desde 36 horas después de la aplicación del parche hasta su retirada a las 72 horas (32). En un estudio de aplicaciones múltiples realizado en voluntarios, la sustitución del parche transdérmico produjo ligeros incrementos en el diámetro pupilar durante un periodo de hasta 12horas, pero permaneció constante hasta la retirada del siguiente parche (23).
. •. En un estudio de aplicación única efectuado en voluntarios, se observaron reducciones dosisdependientes en el diámetro pupilar tras la aplicación de los parches de 35, 52,5 ó 70 µg/l; la isminución del diámetro de la pupila se mantuvo desde 36 horas después de la aplicación del parche hasta su retirada a las 72 horas (32). En un estudio de aplicaciones múltiples. realizado en voluntarios, la sustitución del parche transdérmico produjo ligeros incrementos en el diámetro pupilar durante un periodo de hasta 12horas, pero permaneció constante hasta la retirada del siguiente parche (23).")
31
Perfil farmacocínético
El perfil farmacocinético de buprenorfina, administrada por víaparenteral o sublingual, ha sido previamente revisado en Drugs (17). Los datos publicados acerca de las propiedades farmacocinéticas debuprenorfina administrada mediante el sistema de liberación transdérmico son escasos. Se han realizado dos estudios aleatorizados y abiertos en voluntarios – un estudio de aplicación única (n = 24) (presentado en un póster (33)) y otro de aplicaciones múltiples (n = 54) (datos de archivo (23))- que se han presentado conjuntamente en una monografía (2). Absorción y distribución • Las concentraciones plasmáticas de buprenorfina aumentan de manera sostenida después de la aplicación de un único parche de 35 ó 70 µg/h; las concentraciones mínimas terapéuticamente eficaces (100 pg/ml; sección 1) se alcanzan a las 21 y 11 horas, respectivamente (33). A partir de ese momento, las concentraciones plasmáticas siguieron aumentando hasta alcanzar su máximo (Cmáx) de 305 y 624 pg/ml después de u 60 horas y se mantuvieron por encima de 100 pg/ml hasta el final del periodo de aplicación de 72 horas (23). • Como cabía esperar, el tiempo hasta la Cmáx fue notablementemayor con la administración transdérmica quE durante la infusión intravenosa de 0,3 mg de buprenorfina (0,41 horas), en este estudio cruzado (23). Además, la exposición sistémica a buprenorfina fue mayor con los parches de 35 y 70 µg/h que con la dosis intravenosa, a juzgar por las respectivas áreas bajo la curva concentración-tiempo (AUC media)(20.228, y pg · h/ml) (23). • Las concentraciones plasmáticas de buprenorfina y el AUC aumentan progresivamente con el incremento del número de aplicaciones de parches y parecen alcanzar el estado estacionario después de la tercera aplicación. En un estudio de aplicaciones múltiples (23), los valores medios de la Cmáx de buprenorfina en los voluntarios que recibieron este fármaco (35 µg/h) variaron entre 263,0 y 379,4 pg/ml durante el periodo de observación de 216 horas. Los valores correspondientes a los parches con concentraciones de 52,5 y 70 µg/h fueron de 332,1-528,7
. Los datos publicados acerca de las propiedades farmacocinéticas debuprenorfina administrada mediante el sistema de liberación transdérmico son escasos. Se han realizado dos estudios aleatorizados y abiertos en voluntarios – un estudio de aplicación única (n = 24) (presentado en un póster (33)) y otro de aplicaciones múltiples (n = 54) (datos de archivo (23))- que se han presentado conjuntamente en una monografía (2). Absorción y distribución. • Las concentraciones plasmáticas de buprenorfina aumentan de manera sostenida después de la aplicación de un único parche de 35 ó 70 µg/h; las concentraciones mínimas. terapéuticamente eficaces (100 pg/ml; sección 1) se alcanzan a las 21 y 11 horas, respectivamente (33). A partir de ese momento, las concentraciones plasmáticas siguieron aumentando hasta alcanzar su máximo (Cmáx) de 305 y 624 pg/ml después de u 60 horas y se mantuvieron por encima de 100 pg/ml hasta el final del periodo de aplicación de 72 horas (23). • Como cabía esperar, el tiempo hasta la Cmáx fue notablementemayor con la administración transdérmica quE durante la infusión intravenosa de 0,3 mg de buprenorfina (0,41 horas), en este estudio cruzado (23). Además, la exposición sistémica a buprenorfina fue mayor con los parches de 35 y 70 µg/h que con la dosis intravenosa, a juzgar por las respectivas áreas bajo la curva concentración-tiempo (AUC media)(20.228, y pg · h/ml) (23). • Las concentraciones plasmáticas de buprenorfina y el AUC aumentan progresivamente con el incremento del número de aplicaciones de parches y parecen alcanzar el estado estacionario. después de la tercera aplicación. En un estudio de aplicaciones múltiples (23), los valores medios de la Cmáx de buprenorfina en los voluntarios que recibieron este fármaco (35 µg/h) variaron entre 263,0 y 379,4 pg/ml durante el periodo de observación de 216 horas. Los valores correspondientes a los parches con concentraciones de 52,5 y 70 µg/h fueron de 332,1-528,7.")
32
pg/ml y 390,1-578,2 pglml respectivamente (34)
pg/ml y 390,1-578,2 pglml respectivamente (34). Las pruebas del paso hacia el estado estacionario proceden de la observación de aumentos menos pronunciado del AUC entre la segunda y la terera aplicación del parche (20,34). • Buprenorfina transdérmica tiene una biodisponibilidad de ¾ 50% (35), comparable a la del 50% - 60% tras la administración de buprenorfina sublingual (23). Buprenorfina se une en un 96% a las proteínas plasmáticas (17). Metabolismo y eliminación • En el hígado, buprenorfina se une al ácido glucurónico y se oxida a Ndealquilbuprenorfina (norbuprenorfina) en una reacción mediada por el citocromo P450 (CYP) 3A4 (17, 20, 36). Buprenorfina también se convierte en metabolitos glucuronoconjugados que pueden hidrolizarse en el intestino para formar buprenorfina, más tarde reabsorbida por circulación enterohepática (17, 36). • Buprenorfina tiene una larga semivida de eliminación (t2â). En el estudio de aplicación única, la t2â de buprenorfina 35 y 70 µg/h fue de 25,3 y 27,4 horas, respectivamente, frente a las 8,47 horas para la buprenorfina intravenosa 0,324 mg (23). La t2â en el estudio de aplicaciones múltiples fue de 34,5, 32,6 y 36,8 horas para los parches transdérmicos de 35, 52,5 y 70 g/h, respectivamente (23). Dos tercios de la buprenorfina administrada se excreta en las heces, mientras que el tercio restante lo hace por la orina (17, 36). Interacciones farmacológicas • Dado que buprenorfina se metaboliza en una reacción mediada por el CYP 3A4, la exposición concomitante a fármacos que inhiben o inducen esta enzima puede intensificar o debilitar la acción de buprenorfina (20, 21). La administración simultánea de inhibidores de la monoaminooxidasa, de otros opioides, anestésicos, hipnóticos, sedantes, antidepresivos o neurolépticos puede potenciar los efectos depresivos de buprenorfina sobre el SNC. Asimismo, el alcohol puede intensificar los efectos del fármaco sobre el SNC (20, 21).
. Las pruebas del paso hacia el estado estacionario proceden de la observación de aumentos menos pronunciado del AUC entre la. segunda y la terera aplicación del parche (20,34). • Buprenorfina transdérmica tiene una biodisponibilidad de ¾ 50% (35), comparable a la del 50% - 60% tras la administración de buprenorfina sublingual (23). Buprenorfina se une en un 96% a las proteínas plasmáticas (17). Metabolismo y eliminación • En el hígado, buprenorfina se une al ácido glucurónico y se oxida a Ndealquilbuprenorfina (norbuprenorfina) en una reacción mediada por el citocromo P450 (CYP) 3A4 (17, 20, 36). Buprenorfina también se convierte en metabolitos glucuronoconjugados que pueden hidrolizarse en el intestino para formar buprenorfina, más tarde reabsorbida por circulación. enterohepática (17, 36). • Buprenorfina tiene una larga semivida de eliminación (t2â). En el estudio de aplicación única, la t2â de buprenorfina 35 y 70 µg/h fue de 25,3 y 27,4 horas, respectivamente, frente a las. 8,47 horas para la buprenorfina intravenosa 0,324 mg (23). La t2â en el estudio de aplicaciones múltiples fue de 34,5, 32,6 y 36,8 horas para los parches transdérmicos de 35, 52,5 y 70 g/h, respectivamente (23). Dos tercios de la buprenorfina administrada se excreta en las heces, mientras que el tercio restante lo hace por la orina (17, 36). Interacciones farmacológicas. • Dado que buprenorfina se metaboliza en una reacción mediada por el CYP 3A4, la exposición concomitante a fármacos que inhiben o inducen esta enzima puede intensificar. o debilitar la acción de buprenorfina (20, 21). La administración simultánea de inhibidores de la monoaminooxidasa, de otros opioides, anestésicos, hipnóticos, sedantes, antidepresivos o neurolépticos puede potenciar los efectos depresivos de buprenorfina sobre el SNC. Asimismo, el alcohol puede intensificar los efectos del fármaco sobre el SNC (20, 21).")
33
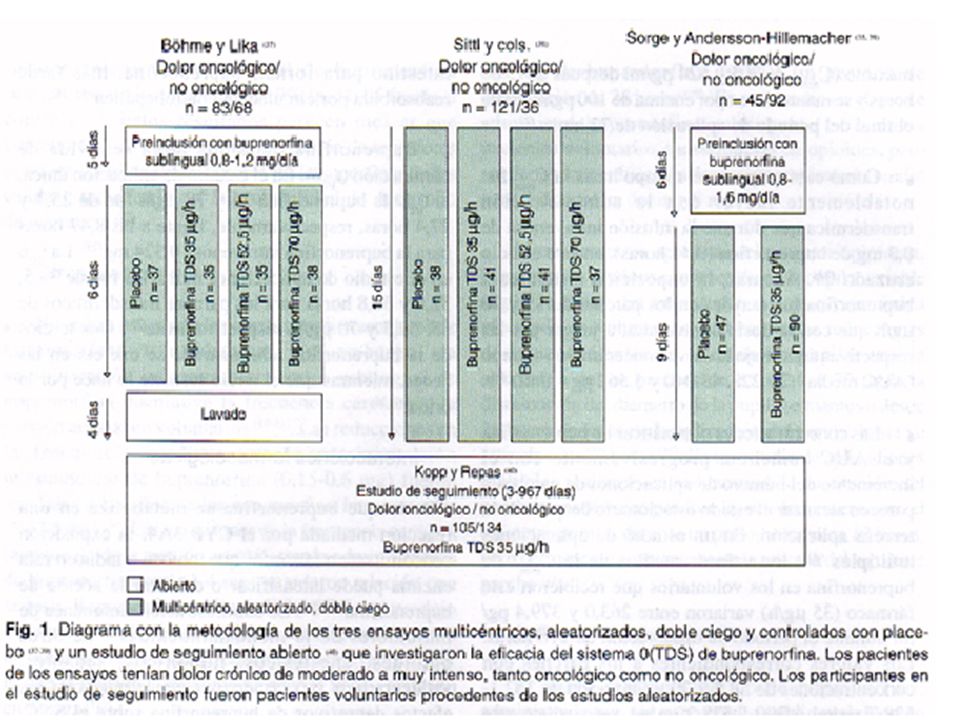
Ensayos terapéuticos La eficacia de los parches de buprenorfina transdérmica en el tratamiento del dolor crónico se ha investigado en tres estudios multicéntricos, aleatorizados, doble ciego, controlados con placebo y de grupos paralelos (37, 39), así como en un estudio de seguimiento abierto realizado en pacientes procedentes de los ensayos aleatorizados que se prestaron voluntarios para seguir recibiendo buprenorfina transdérmica (40) (figura 1). Hasta la fecha, dos de los estudios aleatorizados han sido publicados íntegramente (37, 38) y todos ellos se han comunicado en resúmenes (23, 41-43) y en pósters o comunicaciones (39, 40). En otro informe, se agruparon los datos de los tres ensayos aleatorizados para comparar las medidas del dolor en pacientes con y sin cáncer (44). Los pacientes tenían dolor crónico de moderado a intenso (39) o de intenso a muy intenso (37, 38) tanto oncológico como no oncológico. Los ensayos publicados íntegramente indicaron que 54% de los pacientes eran mujeres y que tenían entre 26 y 86 años (37, 38). Los criterios de exclusión fueron la insuficiencia respiratoria, la hipertensión intracraneal, la hipersensibilidad a los opioides, las lesiones cutáneas extensas preexistentes en la zona de aplicación del parche o la historia de convulsiones (37, 38). También se excluyó a los pacientes en tratamiento con inhibidores de la monoaminooxidasa (37,38), radioisótopos u opioides distintos a la buprenorfina sublingual (37). En dos de los ensayos, se introdujo un periodo de preinclusión (figura 1) durante el cual los pacientes obtuvieron un alivio satisfactorio del dolor mientras recibían buprenorfina sublingual (37, 39). En el otro ensayo, los pacientes que no habían respondido a opioides débiles o a morfina se les cambió de sus analgésicos previos a buprenorfina transdérmica el primer día de tratamiento (38). Los pacientes recibieron al azar buprenorfina transdérmica, en dosis de 35, 52,5 ó 70 µg/h, o placebo, durante un periodo de 6 (37) o 15 (38) días, o recibieron buprenorfina transdérmica, en dosis de 35 µg/h, o placebo, durante 9 días (figura 1) (39). En el estudio de seguimiento, se administró buprenorfina transdérmica, en dosis de 35mg/h, durante días (media: 4,7 meses ) (40). En todos los estudios, los parches transdérmicos se aplicaron en la región subclavicular o la parte superior de la espalda y se sustituyeron cada 72 horas. También se permitió que los pacientes recibieran buprenorfina sublingual como medicación de rescate para el dolor irruptivo (23, 37-40). Cuando así se indicó, serealizaron análisis de eficacia por la población de intención de tratar (37, 38). En los estudios con periodos de tratamiento de 6 a 15 días, el criterio de valoración primario fue el número de pacientes que respondieron (definidos como cualquier paciente en quien el alivio del dolor se consideró al menos satisfactorio en todos los tiempos de evaluación y que recibieron < 0,2 mg/día de buprenorfina sublingual como medicación de rescate a partir del segundo día de tratamiento) (37, 38). En el tercer ensayo aleatorizado, el criterio principal de valoración fue el número de comprimidos sublinguales requeridos durante la segunda y la tercera aplicación del parche, en comparación con el periodo de preinclusión de 6 días (39). El número de tabletas de buprenorfina sublingual (37) o de equivalentes de buprenorfina (38) consumidas durante el periodo de aplicación del parche transdérmico se evaluó como criterio secundario de valoración en los otros dos ensayos aleatorizados. Otros criterios de valoración secundarios a partir de los ensayos aleatorizados incluyeron la evaluación, por el paciente, del alivio del dolor, la intensidad del dolor y la duración del sueño sin dolor (37-39). Sin embargo, estos criterios de valoración secundarios no se analizaron estadísticamente. En el estudio de seguimiento, los criterios de valoración consistieron en el consumo medio diario de comprimidos de buprenorfina sublingual, el alivio del dolor medido sobre una escala de evaluación verbal de cuatro puntos y la facilidad de uso del parche transdérmico(40. 41).
, así como en un estudio de seguimiento abierto realizado en pacientes procedentes de los ensayos aleatorizados que se prestaron voluntarios para seguir recibiendo buprenorfina transdérmica (40) (figura 1). Hasta la fecha, dos de los estudios aleatorizados han sido publicados íntegramente (37, 38) y todos ellos se han comunicado en resúmenes (23, 41-43) y en pósters o comunicaciones (39, 40). En otro informe, se agruparon los datos de los tres ensayos aleatorizados para comparar las medidas del dolor en. pacientes con y sin cáncer (44). Los pacientes tenían dolor crónico de moderado a intenso (39) o de intenso a muy intenso (37, 38) tanto oncológico como no oncológico. Los ensayos publicados íntegramente indicaron que 54% de los pacientes eran mujeres y que tenían entre 26 y 86 años (37, 38). Los criterios de exclusión fueron la insuficiencia respiratoria, la hipertensión intracraneal, la hipersensibilidad a los opioides, las lesiones cutáneas extensas preexistentes en la zona de aplicación del parche o la historia de convulsiones (37, 38). También se excluyó a los pacientes en tratamiento con inhibidores de la monoaminooxidasa (37,38), radioisótopos. u opioides distintos a la buprenorfina sublingual (37). En dos de los ensayos, se introdujo un periodo de preinclusión (figura 1) durante el cual los pacientes obtuvieron un alivio satisfactorio del dolor mientras recibían buprenorfina sublingual (37, 39). En el otro ensayo, los pacientes que no habían respondido a opioides débiles o a morfina se les cambió de sus analgésicos previos a buprenorfina transdérmica el primer día de tratamiento (38). Los pacientes recibieron al azar buprenorfina transdérmica, en dosis de 35, 52,5 ó 70 µg/h, o placebo, durante. un periodo de 6 (37) o 15 (38) días, o recibieron buprenorfina transdérmica, en dosis de 35 µg/h, o placebo, durante 9 días (figura 1) (39). En el estudio de seguimiento, se administró buprenorfina transdérmica, en dosis de 35mg/h, durante días (media: 4,7 meses ) (40). En todos los estudios, los parches transdérmicos se aplicaron en la región subclavicular o la parte superior de la espalda y se sustituyeron cada 72 horas. También se permitió que los pacientes recibieran buprenorfina sublingual como medicación de rescate para el dolor irruptivo (23, 37-40). Cuando así se indicó, serealizaron análisis de eficacia por la población de intención de tratar (37, 38). En los estudios con periodos de tratamiento de 6 a 15 días, el criterio de valoración primario fue el número de pacientes que respondieron (definidos como cualquier paciente en quien el alivio del dolor se consideró al menos satisfactorio en todos los tiempos de evaluación y que recibieron < 0,2 mg/día de buprenorfina sublingual como medicación de rescate a partir del segundo día de tratamiento) (37, 38). En el tercer ensayo aleatorizado, el criterio principal de valoración fue el número de comprimidos sublinguales requeridos durante la segunda y la tercera aplicación del parche, en comparación con el periodo de preinclusión de 6 días (39). El número de tabletas de buprenorfina sublingual (37) o de equivalentes de buprenorfina (38) consumidas durante el periodo de aplicación del parche transdérmico se evaluó como criterio secundario de valoración en los otros dos ensayos aleatorizados. Otros criterios de valoración secundarios a partir de los ensayos aleatorizados incluyeron la evaluación, por el paciente, del alivio del dolor, la intensidad del dolor y la duración del sueño sin dolor (37-39). Sin embargo, estos criterios de valoración secundarios no se analizaron estadísticamente. En el estudio de seguimiento, los criterios de valoración consistieron en el consumo medio diario de comprimidos de buprenorfina sublingual, el alivio del dolor medido sobre una escala de evaluación verbal de cuatro puntos y la facilidad de uso del parche transdérmico(40. 41).")
34
• Al menos un tercio de los pacientes tratados con buprenorfina transdérmica respondieron al tratamiento en dos ensayos aleatorizados (figura 2) (37, 38). Cabe destacar que > 33% de los tratados sin éxito con opioides débiles o morfina respondieron al tratamiento con buprenorfina transdérmica y que el porcentaje de pacientes con respuesta en los grupos de tratamiento con 35 y 52,5 gg/h fue significativamente mayor que en los que recibieron placebo en este estudio (p ¡Ü0,032) (38). Sin embargo, no se observó ningún incremento en la respuesta con el parche de 70 µg/h (figura 2), lo que se atribuyó al número de pacientes refractarios en este grupo de tratamiento (38). • De los pacientes que tuvieron alivio del dolor satisfactorio con buprenorfina sublingual durante la fase de preinclusión de 5 días, del 34% al 50% respondieron, en función de la dosis, a un tratamiento de 6 días con buprenorfina transdérmica (37), Aunque se observó una diferencia con placebo, no alcanzó significación estadística (figura 2). Un análisis retrospectivo de subgrupos de los resultados de los tres ensayos aleatorizados revelaron una respuesta al tratamiento tanto en pacientes con dolor oncológico como en pacientes sin cáncer (44). • La necesidad de medicación de rescate (buprenorfina sublingual) disminuyó, respecto al periodo basal, en aproximadamente el 50%-70% en pacientes tratados con buprenorfma transdérmica (35, 45). En los casos con alivio satisfactorio del dolor tras un periodo de preinclusión de 5 días (37), el consumo de buprenorfina sublingual en los grupos de tratamiento activo disminuyó, en función de la dosis (presentado en un póster (45), véase la figura 3). En pacientes con dolor insuficientemente controlado con opioides débiles o morfina, la necesidad de la administración adicional de analgésicos opioides orales se redujo en >50% con buprenorfina transdérmica. Para los grupos de tratamiento activo todas la reducciones en la necesidad de medicación de rescate fueron significativamente mayores (p< 0,05) que las observadas en los que recibieron placebo (8%; figura 3) (38).
. Un análisis retrospectivo de subgrupos de los resultados de los tres ensayos aleatorizados revelaron una respuesta al tratamiento tanto en pacientes con dolor oncológico como en pacientes sin cáncer (44). • La necesidad de medicación de rescate (buprenorfina sublingual) disminuyó, respecto al periodo basal, en aproximadamente el 50%-70% en pacientes tratados con buprenorfma. transdérmica (35, 45). En los casos con alivio satisfactorio del dolor tras un periodo de preinclusión de 5 días (37), el consumo de buprenorfina sublingual en los grupos de tratamiento activo. disminuyó, en función de la dosis (presentado en un póster (45), véase la figura 3). En pacientes con dolor insuficientemente controlado con opioides débiles o morfina, la necesidad. de la administración adicional de analgésicos opioides orales se redujo en >50% con buprenorfina transdérmica. Para los grupos de tratamiento activo todas la reducciones en la necesidad de medicación de rescate fueron significativamente mayores (p< 0,05) que las observadas en los que recibieron placebo (8%; figura 3) (38).")
35
• La necesidad de medicación de rescate también se relacionó con la necesidad de buprenorfina sublingual en el periodo basal (39). En el estudio que comparó buprenorfina transdérmica 35 µg/h con placebo se observó que en pacientes que recibían 0,4-0,8, 0,8-1,2, 1,2-1,6 ó ¡Ý 1,6 mg de buprenorfina sublingual durante el periodo de preinclusión, las necesidades de medicación de rescate durante el periodo de tratamiento de buprenorfina transdérmica fueron de 0,2, 0,4, 0,6 y 0,9 mg, respectivamente (39). Cuando se combinó la cantidad de buprenorfina utilizada como medicación de rescate con la dosis diaria administrada mediante el parche de buprenorfina transdérmica (0,8 mg), la dosis total de buprenorfma administrada fue casi exactamente igual a la dosis recibida durante el periodo de preinclusion (42). • A pesar de la disponibilidad de medicación de rescate para todos los pacientes, los que recibieron buprenorfina transdérmica tendieron a experimentar un mayor alivio del dolor que los tratados con placebo (37-39). En uno de los ensayos, el porcentaje de pacientes que manifestaron un alivio del dolor de bueno a total aumentó hasta en un 13,2% y 8,6% durante la aplicación del primer y segundo parche de buprenorfina transdérmica, respectivamente, frente a una reducción del 20% y el 11,4% en los pacientes que recibieron placebo (37). Durante los 15 días de tratamiento, el 40,4%-46,3% de los pacientes tratados con buprenorfina transdérmica tuvieron un alivio del dolor entre bueno y total en comparación con el 32,4% de los que recibieron placebo(38). Esto también se reflejó en la media de las puntuaciones del alivio del dolor registradas por los pacientes durante el estudio; las medias de las puntuaciones verbales de evaluación (determinadas mediante una escala de cuatro puntos [1-4: escaso, satisfactorio, bueno o total]) fueron de 2,3, 2,4 y 2,5 en los pacientes tratados con buprenorfma transdérmica 35, 52,5 y 70 µg/h, respectivamente, frente a 1,9 en los que recibieron placebo (38). •
. Cuando se combinó la cantidad de buprenorfina utilizada como medicación de rescate con la dosis diaria administrada mediante el parche de buprenorfina transdérmica (0,8 mg), la dosis total de buprenorfma administrada fue casi exactamente igual a la dosis recibida durante el periodo de preinclusion (42). • A pesar de la disponibilidad de medicación de rescate para todos los pacientes, los que recibieron. buprenorfina transdérmica tendieron a experimentar un mayor alivio del dolor que los tratados con placebo (37-39). En uno de los ensayos, el porcentaje de pacientes que manifestaron un alivio del dolor de bueno a total aumentó hasta en un 13,2% y 8,6% durante la aplicación del primer y segundo parche de buprenorfina transdérmica, respectivamente, frente a una reducción del 20% y el 11,4% en los pacientes que recibieron placebo (37). Durante los 15 días de tratamiento, el 40,4%-46,3% de los pacientes tratados con buprenorfina transdérmica tuvieron un alivio del dolor entre bueno y total en comparación con el 32,4% de los que. recibieron placebo(38). Esto también se reflejó en la media de las puntuaciones del alivio del dolor registradas por los pacientes durante el estudio; las medias. de las puntuaciones verbales de evaluación (determinadas mediante una escala de cuatro puntos [1-4: escaso, satisfactorio, bueno o total]) fueron de 2,3, 2,4 y 2,5 en los pacientes tratados con buprenorfma transdérmica 35, 52,5 y 70 µg/h, respectivamente, frente a 1,9 en los que recibieron placebo (38). •")
36
En el estudio de 15 días, se observó un incremento, en función de la dosis, de la proporción de pacientes sin dolor o con dolor leve, en cada día del estudio y al final del mismo (38). En el último día de tratamiento, el 46,3%, el 60,0% y el 61,1% de los pacientes tratados con buprenorfina, en dosis de 35, 52,5 y 70 µg/h, respectivamente, experimentaron dolor leve o nulo, en comparación con el 40,5% de los que recibieron placebo (38). Además, el último día de estudio el 17%, 7,5% y 13,9% de los pacientes tratados con buprenorfina transdérmica manifestaron dolor intenso y muy intenso, frente al 24,3% de los que recibieron placebo (38). • La proporción de pacientes con > 6horas de sueño sin dolor por noche aumentó en los tratados con buprenorfina (incremento del 2,5%- 11,9% respecto al periodo basal) y disminuyó en los que recibieron placebo (reducción del 15,3% y el 5,9%) durante 6 (37) o 9 (39) días de tratamiento.Durante la fase de tratamiento de dos ensayos, el 40%-55% de los tratados con buprenorfina transdérmica tuvieron un sueño que no fue interrumpido por el dolor, en comparación con aproximadamente el 35% de los que recibieron placebo (37, 38).
. • La proporción de pacientes con > 6horas de sueño sin dolor por noche aumentó en los tratados con buprenorfina (incremento del 2,5%- 11,9% respecto al periodo basal) y disminuyó en los que recibieron placebo (reducción del 15,3% y el 5,9%) durante 6 (37) o 9 (39) días de tratamiento.Durante la fase de tratamiento de dos ensayos, el 40%-55% de los tratados con buprenorfina transdérmica tuvieron un sueño que no fue interrumpido por el dolor, en comparación con aproximadamente el 35% de los que recibieron placebo (37, 38).")
Presentaciones similares










![La eficacia de la condroitina en el tratamiento de la artrosis es dudosa AP al día [ ] Reichenbach.](/12/3380941/big_thumb.jpg "La eficacia de la condroitina en el tratamiento de la artrosis es dudosa AP al día [ ] Reichenbach.>")










