Descargar la presentación
La descarga está en progreso. Por favor, espere

1
CURSO SOBRE PREVENCIÓN DE DEFICIENCIAS
Alcañiz. Enero, 2009 PREVENCIÓN DE ERRORES CONGÉNITOS DEL METABOLISMO Dr. Antonio Baldellou Unidad de Enfermedades Metabólicas Hospital Infantil Miguel Servet. Zaragoza

3
MPS I patients visit a variety of specialists (paediatricians most frequently) before a final diagnosis is made
 before a final diagnosis is made")
4
Long interval between first symptoms and diagnosis in Fabry disease

5
PREVENCIÓN DE ERRORES CONGÉNITOS
DEL METABOLISMO Introducción Concepto y clasificación de ECM Carga sanitaria de los ECM Estrategias de prevención de las ECM Conclusiones

6
I. Introducción

7
Pitágoras de Samos Siglo VI A.C. Prohíbe la ingesta de habas

8
Sir Archibald E. Garrod Año 1908 Royal College of Physicians of London Concepto de error congénito del metabolismo

10
Bickel H Influence of phenylalanine intake on phenylketonuria Lancet, 1953; II:

11
Robert Guthrie A simple phenylalanine method for detection phenylketonuria in large populations of newborn infants Pediatrics 1963; 32:

12
“…Every patient who has on of the so-called “single-gene” diseases described in this book has one “orphan” disease: furthemore, because of biological identity, each patient has his or her own private (orphan) form of unhealth. In other words, this book is an ultimate guidebook for individualized medicine…” Charles R. Scriver, MMBID-8, Preface
 form of unhealth. In other words, this book is an ultimate guidebook for individualized. medicine… Charles R. Scriver, MMBID-8, Preface.")
13
II. Concepto y clasificación de ECM

14
CONCEPTO DE ERROR CONGÉNITO DEL METABOLISMO
Los errores congénitos del metabolismo son trastornos bioquímicos determinados genéticamente, de la función o estructura de una proteína.

15
MUTACIÓN GÉNICA ALTERACIÓN ESTRUCTURA O FUNCIÓN DE UNA PROTEÍNA FACTORES GÉNICOS FACTORES EPIGENÉTICOS ALTERACIÓN HOMEOSTASIS ALTERACIÓN ORGÁNICA SÍNTOMAS CLÍNICOS ENFERMEDAD METABÓLICA

16
E.C.M. CLASIFICACIÓN I. Alteraciones del metabolismo intermediario II. Alteraciones del metabolismo de las moléculas complejas III. Otros…

17
Alteraciones del metabolismo intermediario

18
ALTERACIONES DEL METABOLISMO INTERMEDIO
Trastornos del metabolismo de los aminoácidos Alteraciones de la -oxidación de los ácidos grasos y de la cetogénesis Trastornos del transporte y metabolismo de los carbohidratos Alteraciones mitocondriales Alteraciones congénitas del metabolismo de las vitaminas Alteraciones del transporte de aminoácidos Trastornos del metabolismo de los péptidos Alteraciones del metabolismo de los minerales

19
ALTERACIONES DEL METABOLISMO DE LAS MOLÉCULAS COMPLEJAS

20
ALTERACIONES DEL METABOLISMO DE MOLÉCULAS
COMPLEJAS Alteraciones lisosomales Alteraciones peroxisomales Trastornos del metabolismo de esteroles Trastornos del metabolismo de los ácidos biliares y del heme Anomalías congénitas de la glicosilación Trastornos del metabolismo de las lipoproteínas

21
III. Carga sanitaria de los ECM

22
FRECUENCIA DE LAS ENFERMEDADES METABÓLICAS
Uno de cada 800 – recién nacidos nace afecto de un error congénito del metabolismo Casi dos tercios de ellos dan lugar a un conjunto de signos y síntomas que configuran una “enfermedad metabólica congénita” El 80 % de estas enfermedades debuta durante la infancia El 50 % lo hace durante la época del recién nacido

24
ENFERMEDADES METABÓLICAS ÓRGANOS Y SISTEMAS AFECTOS
ÓRGANO / SISTEMA % AFECTACIÓN SNC Sangre Hígado-Páncreas 17 Esqueleto 16 Ojos Piel Genitourinario 15 Endocrino 14 Circulatorio 14 Muscular Inmunológico 12 ORL 5 Jimenez-Sánchez G, MMBID-8

25
COSTE SANITARIO DE UN PACIENTE AFECTO DE UN ECM
Gasto medio: – € /vida

26
IV. Estrategia de prevención de las ECM

27
PREVENCIÓN DE LAS E.C.M. Primaria Asesoramiento familiar Diagnóstico preimplantacion Secundaria Diagnóstico prenatal Examen sistemático del recién nacido Diagnóstico precoz ► clínico ► bioquímico ► molecular Terciaria Tratamiento

28
PREVENCIÓN PRIMARIA DE LAS E.C.M.
ASESORAMIENTO FAMILIAR 60 % de las EM tienen herencia AR 20 % de las EM tienen herencia AD 20 % de las EM tienen herencia X-L o Mitocondrial DIAGNÓSTICO PREIMPLANTACION

29
PREVENCIÓN PRIMARIA DE LAS E.C.M.
VENTAJAS Evita la aparición de la enfermedad INCONVENIENTES Habitualmente se inicia tras la aparición de un afecto DP técnicamente complejo Asesoramiento familiar probabilístico

30
PREVENCIÓN SECUNDARIA DE LAS E.C.M.
1. DIAGNÓSTICO PRENATAL Selección de sexo Diagnóstico durante el embarazo ► molecular ► bioquímico / enzimático ► morfológico

31
DIAGNÓSTICO PRENATAL Ventajas Fiabilidad técnica creciente Inconvenientes Caso índice debe estar perfectamente definido Procedimientos diagnósticos limitados en el tiempo Procedimientos diagnósticos complejos Muestras delicadas y de transporte comprometido Problemas éticos no siempre fáciles de resolver

32
PREVENCIÓN SECUNDARIA DE LAS E.C.M
2. DIAGNÓSTICO SISTEMÁTICO NEONATAL POBLACIÓN NO SELECCIONADA POBLACION SELECCIONADA a. etnia: mediterráneos, raza negra, gitanos, canadienses askenazis, irlandeses, etc. b. antecedentes familiares

33
E.C.M. DIAGNÓSTICO NEONATAL SCREENING INTERVENCIÓN DEL PEDIATRA
CLÁSICO Hiperfenilalaninemias Todos los demás Hipotiroidismos Hiperplasia cong. suprarrenal F.Q.P. “AMPLIADO” MS/MS Alteraciones metabolismo aminoácidos Alteraciones ciclo de la urea Defectos de la b-oxidación Enfermedades lisosomales Micro “chips” Enfermedades con mutaciones identificadas

34
I. UTILIDAD DEL MÉTODO DE SECREENING MS/MS
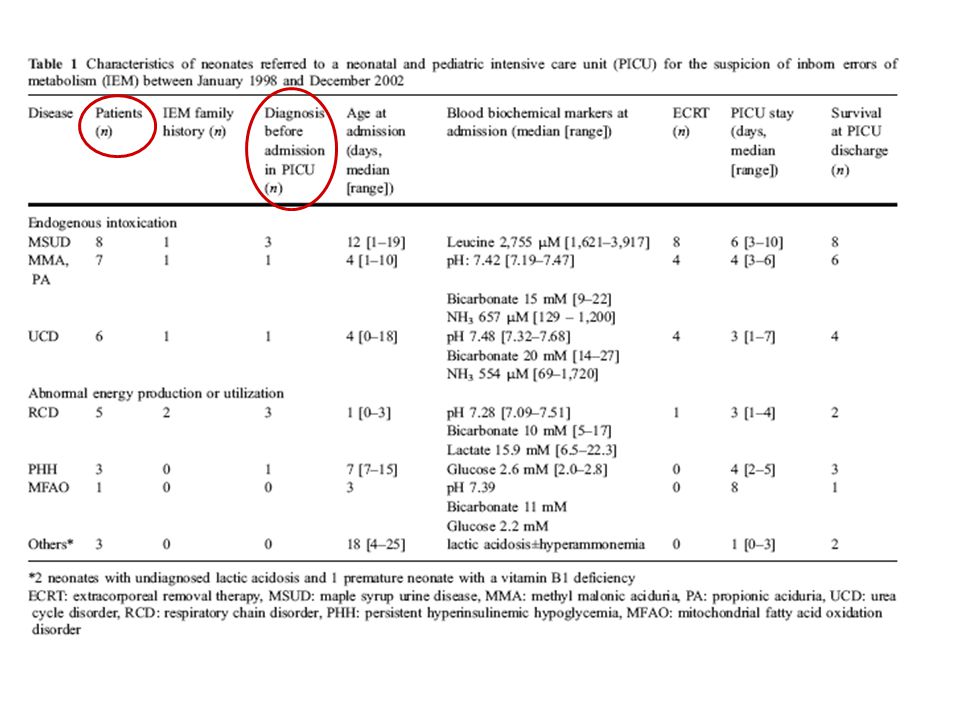
COSTES Por proceso € Por paciente diagnosticado € Coste anual por paciente no identificado por screening € *Antes de la implantación del cribado neonatal ampliado se diagnosticaba (en Galicia) una enfermedad metabólica por cada recién nacidos, y tras su introducción de ha diagnosticado un caso por cada recién nacidos.
 una enfermedad metabólica por. cada recién nacidos, y tras su introducción de ha diagnosticado un caso por cada recién nacidos.")
35
POBLACIONES SELECCIONADAS
POBLACIÓN ENFERMEDAD DE RIESGO Askenazis Tay-Sachs Norrborttian Gaucher III Canadienses Tirosinemia I Mediterráneos Def. glucosa-6-fosfato deshidrogenasa -talasemia Gitanos MCAD Raza negra Hemoglobinopatías Menonitas MSUD Irlandeses Homisctinuria (CBS) Daneses
 Daneses.")
36
DIAGNÓSTICO SISTEMÁTICO NEONATAL
Ventajas Nuevas tecnologías ►ampliación de las posibilidades diagnósticas Se trata del método diagnóstico “ideal” sencillo sensible y específico precoz no “debería” estar sometido a fallos humanos Puede poner en evidencia alteraciones maternas desapercibidas Inconvenientes Sigue teniendo un alcance limitado Su progresiva expansión plantea problemas diagnósticos difíciles Los falsos negativos pueden tener consecuencias muy lamentables Los falsos positivos pueden originar una gran carga emocional familiar Su progresiva ampliación genera carga económica creciente

37
PREVENCIÓN SECUNDARIA DE LAS E.C.M 3.1 . DIAGNOSTICO CLÍNICO
A. Antecedentes familiares Consanguinidad Fallecimientos inexplicados en la familia Antecedentes familiares conocidos 60 % de las EM tienen herencia AR 20 % de las EM tienen herencia AD 20 % de las EM tienen herencia X-L o Mitocondrial

38
ENFERMEDADES METABÓLICAS DIAGNOSTICO CLINICO
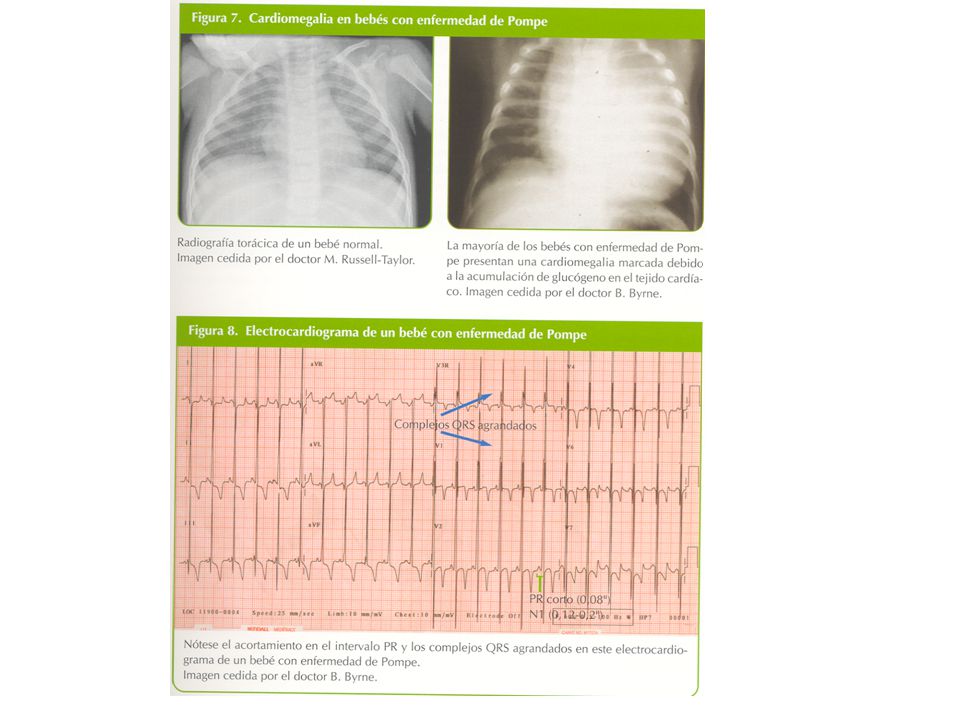
B. SINTOMATOLOGÍA I. 1. PATOLOGÍA DEL METABOLISMO INTERMEDIO INTOXICACIONES ENTIDADES CARACTERÍSTICAS GENERALES Alt. Aminoácidos Intérvalo libre Ac. Orgánicas Síntomas “tóxicos”agudos o crónicos Def. ciclo urea Trastornos metabólicos recurrentes Intolerancia azúcares La clínica puede ser tardía o intermitente B DÉFICITS ENERGÉTICOS Glucogenosis Variabilidad en el inicio de los síntomas Def. gluconeogénesis Síntomas de insuficiencia hepática, Acidosis lácticas muscular, cardiaca, del SNC, etc. Def. oxidación de las grasas Muerte súbita infantil OXPHOS Malformaciones congénitas frecuentes

39
HIPERGLICINEMIA NO CETÓSICA

41
ENFERMEDADES METABÓLICAS DIAGNOSTICO CLINICO
B. SÍNTOMATOGÍA I. 2. PATOLOGÍA DE LAS MOLÉCULAS COMPLEJAS ENTIDADES CARACTERÍSTICAS GENERALES Lisosomales Síntomas permanentes Peroxisomales Síntomas progresivos de afectación orgánica Deficiencia de A1AT Fenotipos malformativos y dismórficos Alteración síntesis de colesterol característicos Al. de síntesis de ácidos biliares CDG I. 3. OTRAS ENFERMEDADES Alt. Purinas y pirimidinas Síntomas muy variados Porfirias Frecuente afectación neurológica Alt. Metabolismo creatina Alt. Metabolismo Cu, Zn, Fe Alteraciones del transporte Etc.

42
MPS I MANIFESTACIONES CLÍNICAS Evolución crónica y progresiva Afectación multisistémica Organomeglia Disóstosis múltiple Hernia umbilical/inguinal Facies “tosca” Artropatía severa Afectación de la audición Afectación de la visión Afectación respiratoria Afectación cardiovascular

43
DIAGNÓSTICO CLINICO Ventajas El sistema sanitario español facilita la sospecha diagnóstica acceso universal y gratuito programas de salud infantil control del niño por el Pediatra Inconvenientes Depende de la calidad del acto médico conocimientos suficientes ► aplicados a la asistencia sanitaria

44
DIAGNÓSTICO ENFERMEDADES METABÓLICAS
3.2. DIAGNÓSTICO BIOQUÍMICO Identificación de alteraciones bioquímicas Identificación de alteraciones enzimáticas

45
DIAGNÓSTICO ENFERMEDADES METABÓLICAS
DIAGNÓSTICO BIOQUÍMICO Ventajas Los avances metodológicos agigantan las posibilidades diagnósticas El transporte de las muestra biológicas no suele ser difícil Inconvenientes Técnicas complejas en muchas ocasiones Precisan orientación clínica previa Es necesario saber interpretar adecuadamente los resultados

46
COMPROBACIÓN FENOTIPO BIOQUÍMICO COMPATIBLE
MUESTRA PRACTICAR CONSERVAR Sangre Glucosa Dry Spot Gases. Anión GAP 10 c.c. Sangre EDTA (- 20º) Ac. Úrico 3 ml plasma Amonio Láctico / Pirúvico 3OHButírico / Acetoacético Ácidos grasos libres Aminoácidos Pruebas coagulación Hemograma Orina C. Cetónicos Muestras antes y después C. Reductores de cualquier tratamiento. Test Sulfitos y Brand pH, Ionograma Ac. Úrico Otras ml LCR (- 20º) Cultivo fibroblastos
 Ac. Úrico 3 ml plasma. Amonio. Láctico / Pirúvico. 3OHButírico / Acetoacético. Ácidos grasos libres. Aminoácidos. Pruebas coagulación. Hemograma. Orina C. Cetónicos Muestras antes y después. C. Reductores de cualquier tratamiento. Test Sulfitos y Brand. pH, Ionograma. Ac. Úrico. Otras 2 ml LCR (- 20º) Cultivo fibroblastos.")
47
DIAGNÓSTICO ENFERMEDADES METABÓLICAS
3.3. DIAGNÓSTICO MOLECULAR ADN NUCLEAR Frecuencia de anomalías: 1 / 800 – recién nacidos 2. ADN MITOCONDRIAL Frecuencia de anomalías : 1 / recién nacidos

48
DIAGNÓSTICO ENFERMEDADES METABÓLICAS DIAGNÓSTICO MOLECULAR
ADN NUCLEAR Ventajas Puede utilizarse cualquier célula nucleada Diagnóstico preciso Ayuda a “definir” al paciente en bastantes ocasiones Permite información génica familiar adecuada Simplifica el diagnóstico prenatal Inconvenientes Técnicas complejas y largas en muchas ocasiones No siempre se identifican las mutaciones patógenas ADN MITOCONDRIAL Su tamaño reducido facilita su estudio Todos los tejidos no son iguales para su estudio Es muy difícil interpretar los resultados No es fácil una correlación genotipo/fenotipo Su uso en el diagnóstico prenatal es todavía muy limitado

49
PREVENCION TERCIARIA DE LAS ECM
POSIBILIDADES TERAPÉUTICAS TRATAMIENTO DEL FENOTIPO CLÍNICO TRATAMIENTO DEL PERFIL BIOQUÍMICO TRATAMIENTO DE LA ESTRUCTURA O FUNCIÓN DE UNA PROTEÍNA TRATAMIENTO GÉNICO

50
TRATAMIENTO EN LAS ENFERMEDADES METABÓLICAS
TRATAMIENTO DEL FENOTIPO CLÍNICO Cirugía malformaciones Ortopedia alteraciones esqueléticas Tratamiento farmacológico anticonvulsivantes bifosfonatos Etc. Trasplante de órganos sustitución de órganos dañados riñón (cistinosis, oxaluria ) hígado (Wilson, α-1-AT, Niemann-Pick, etc..) corazón (FQP, defectos -oxidación, etc )
 hígado (Wilson, α-1-AT, Niemann-Pick, etc..) corazón (FQP, defectos -oxidación, etc )")
51
TRATAMIENTO EN LAS ENFERMEDADES METABÓLICAS
2. TRATAMIENTO DEL FENOTIPO BIOQUÍMICO Restricción ingesta sustrato alteraciones metabolismo aminoácidos, ciclo urea Bloqueo de la absorción intestinal del sustrato uso de inhibidores de la absorción de colesterol Inhibición de la síntesis de sustratos “iminoazúcares” para inhibir acúmulo de glicoesfingolípidos dicloroacetato en los defectos de PDH alopurinol en la hiperuricemia NTBC en la tirosinemia I Eliminación-quelación del sustrato benzoato sódico / fenilbutirato en el ciclo urea Bloqueo de los receptores celulares dextrometorfan en la hiperglicinemia no cetósica Puesta en marcha de vías metabólicas alternativas cisteamina en la cistinosis Administración del producto deficiente colesterol en el S-L-O

52
TRATAMIENTO EN LAS ENFERMEDADES METABÓLICAS
3. TRATAMIENTO DE LA ESTRUCTURA / FUNCIÓN DE UNA PROTEÍNA Uso de vitaminas como coenzimas Vitaminas C, fólico, B1, B2, niacina, B6, B12, K1 Administración de cofactores Deficiencias de síntesis o regeneración de BH4 Estabilización enzimática Uso de chaperonas Estimulación enzimática “Carbaglu” en defectos del ciclo de la urea Administración de la proteína deficiente (T.E.S.) Gaucher, MPS I, Fabry MPS II, VI, Pompe MPS IV, VII, Niemann Pick B Trasplante de órgano como fuente de células productoras de enzima Hígado: Ciclo urea, MSUD, etc. Trasplante de células Hepatocitos Trasplante de médula ósea o HSCT como fuente de células “madre” X-ALD, MLD, Gaucher III, Osteopetrosis, MPS I, etc..
 Gaucher, MPS I, Fabry. MPS II, VI, Pompe. MPS IV, VII, Niemann Pick B. Trasplante de órgano como fuente de células productoras de enzima. Hígado: Ciclo urea, MSUD, etc. Trasplante de células. Hepatocitos. Trasplante de médula ósea o HSCT como fuente de células madre X-ALD, MLD, Gaucher III, Osteopetrosis, MPS I, etc..")
53
TRATAMIENTO EN LAS ENFERMEDADES METABÓLICAS
4. TRATAMIENTO GÉNICO Trasplante de órganos o de células no modificadas trasplante de órganos trasplante auxiliar de órganos trasplante de células Implante de células modificadas “ex vivo” células del paciente líneas celulares alogénicas Administración de genes mediante vector vector plasmídico vector viral vector no viral Manipulación de la expresión génica Inhibición de la síntesis del ADN anormal

54
V. CONCLUSIONES

55
EL ADECUADO CONTROL DE LAS ENFERMEDADES METABÓLICAS,
REQUIERE: Red sanitaria adecuada personal sanitario recursos técnicos Coordinación estatal Apoyo a las Asociaciones de familiares

Presentaciones similares



>")






 ¿ENFERMEDADES RARAS?>")










