Descargar la presentación
La descarga está en progreso. Por favor, espere

1
CURSO DE FARMACOLOGIA I
Dr. Honorio R. Pérez Martínez

2
Biofarmacia. Farmacocinética. Absorción de un fármaco, biodisponibilidad, cinética, distribución, unión a proteínas plasmáticas

3
La biofarmacia es la rama de la farmacología que se encarga del estudio de la influencia de la forma y la formulación química y física de un medicamento sobre los acontecimientos farmacocinéticos y farmacodinámicos consecutivos a su administración

4
Biofarmacia estudia la influencia de la formulación y la técnica de elaboración de un medicamento sobre su actividad terapéutica. En ella se consideran los efectos de la forma de dosificación sobre la respuesta biológica y los factores que pueden afectar al principio activo y a la forma farmacéutica que lo incluye

5
El interés básico del estudio de estas materias se basa en determinar las dosis más adecuadas y el intervalo de administración en formas farmacéuticas de biodisponibilidad óptima.

6
Por otra parte, permite predecir y calcular la concentración de los fármacos en diferentes
órganos, con el fin de instaurar un régimen terapéutico óptimo.

7
El conjunto de procesos que caracterizan la evolución temporal de un medicamento, tras ser administrado a un organismo, en determinadas condiciones y bajo una vía de administración específica, se denomina LADME

8
FARMACOCINÉTICA. Estudia qué hace el organismo cuando recibe un fármaco, ó sea, los cambios que el organismo le hace a los medicamentos; son características farmacocinéticas de los medicamentos, la liberación, absorción, distribución, metabolismo y excreción.

9
Este campo de la Farmacología está regulado por leyes expresadas en modelos matemáticos.
Es muy importante para predecir la acción terapéutica ó tóxica de los fármacos

10
FARMACODINÁMICA. Estudia las acciones y los efectos que los fármacos producen en los distintos órganos y sistemas del cuerpo.

11
IMPORTANCIA SOCIOSANITARIA DE LOS MEDICAMENTOS
-Prolongación de la calidad y esperanza de vida con el descubrimiento y aplicación de nuevos medicamentos -Reto de enfermedades nuevas que necesitan desarrollar nuevos medicamentos, ó la resistencia a los antibióticos

12
-El medicamento No debe tratarse como un bien de consumo más, sino ir orientado a lograr el bienestar y la salud del paciente y la población -Uso racional de medicamentos (antibióticos, analgésicos, etc.)
")
13
-Control del uso, en pacientes mayores de 65 años
-Educación sanitaria a la población, eficaz para controlar los efectos adversos y hacer un uso racional de los medicamentos

14
-Riesgo de que la industria farmacéutica produzca “nuevos medicamentos”, que no ofrezca ninguna ventaja sobre el que existía

15
FARMACINETICA Estudia el movimiento de los fármacos en el organismo y permite conocer su concentración en la Biofase. Para que un fármaco pueda ejercer su acción debe alcanzar una concentración crítica en la BIOFASE.

16
BIOFASE. Es el medio en el cual el fármaco está en condiciones de interactuar con sus receptores para ejercer su efecto biológico, ya sea terapéutico ó tóxico

17
Para alcanzar esta concentración crítica es necesario que el fármaco cumpla estos procesos: (LADME)
1-Se libere desde su formulación farmacéutica (L) 2-Penetre en el organismo a través de los procesos de absorción (A)
 2-Penetre en el organismo a través de los procesos de absorción (A)")
18
3-Llegue al plasma y por medio de este a los tejidos a través de un proceso de distribución (D)
4-Tan pronto como penetra al organismo está sometido a mecanismos de eliminación, que pueden ser por vías naturales (orina, bilis, saliva, etc.) o por sufrir una biotransformación ó metabolismo (M) (E)
 o por sufrir una biotransformación ó metabolismo (M) (E)")
19
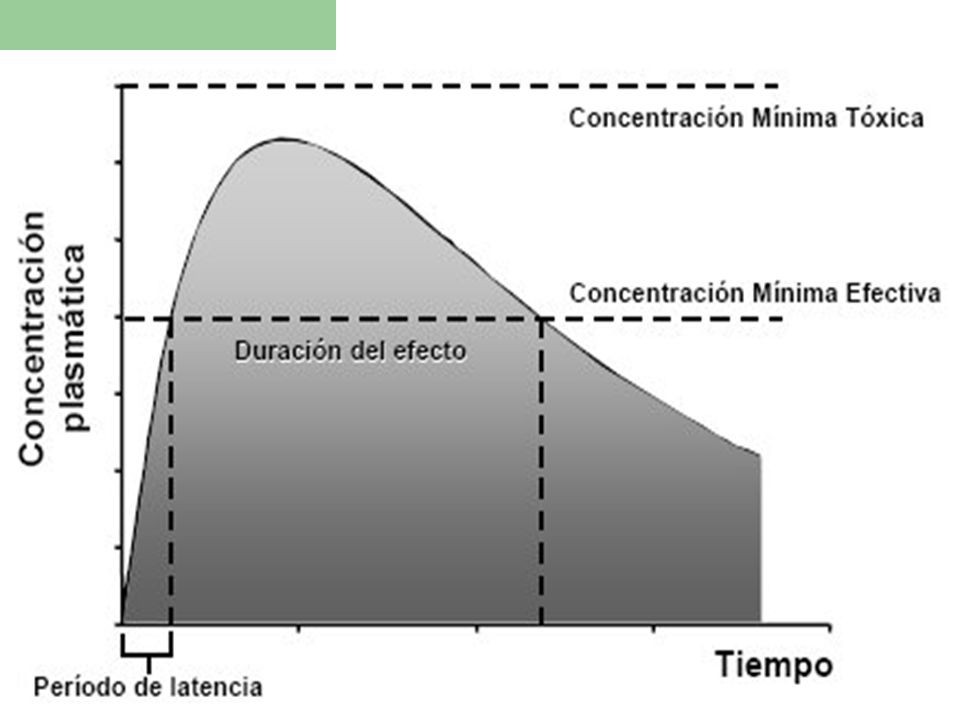
CURVA DE NIVELES PLASMATICOS
En la práctica resulta muy difícil medir las concentraciones tisulares del fármaco en la biofase, pero si es posible hacerlo por los niveles obtenidos en el plasma de un fármaco, para predecir un efecto Esta curva describe las variaciones sufridas por la concentración del fármaco en el plasma desde su administración hasta su desaparición del organismo

20
Parámetros Importantes que se observan en ésta curva:
a-Concentración Mínima Eficaz (CME) o Terapéutica. Es aquella concentración a partir de la cual se inicia el efecto farmacológico o terapéutico
 o Terapéutica. Es aquella concentración a partir de la cual se inicia el efecto farmacológico o terapéutico.")
21
b-Concentración Mínima Tóxica (CMT)
b-Concentración Mínima Tóxica (CMT). Es aquella concentración a partir de la cual suelen aparecer o iniciarse los efectos tóxicos. c-Índice Terapéutico o “margen de seguridad”. Es el cociente entre la CMT y CME; cuanto mayor es esta relación, mayor seguridad ofrece la administración de un fármaco
. Es aquella concentración a partir de la cual suelen aparecer o iniciarse los efectos tóxicos. c-Índice Terapéutico o margen de seguridad . Es el cociente entre la CMT y CME; cuanto mayor es esta relación, mayor seguridad ofrece la administración de un fármaco.")
23
d-Período de Latencia (PL)
d-Período de Latencia (PL). Tiempo transcurrido desde la administración hasta que se alcanza la CME.
. Tiempo transcurrido desde la administración hasta que se alcanza la CME.")
24
e-Intensidad del Efecto (IE)
e-Intensidad del Efecto (IE). Este aspecto tiene relación con la concentración alcanzada en el plasma. Sin embargo hay que tener en cuenta que el paso del fármaco de la sangre a los tejidos depende de algunos factores, tales como: unión a las proteínas del plasma, ya que sólo la fracción libre de este difunde a los tejidos, el flujo sanguíneo de cada tejido, y la afinidad del fármaco por un tejido determinado
. Este aspecto tiene relación con la concentración alcanzada en el plasma. Sin embargo hay que tener en cuenta que el paso del fármaco de la sangre a los tejidos depende de algunos factores, tales como: unión a las proteínas del plasma, ya que sólo la fracción libre de este difunde a los tejidos, el flujo sanguíneo de cada tejido, y la afinidad del fármaco por un tejido determinado.")
25
f-Duración de la acción ó Tiempo Eficaz (TE)
f-Duración de la acción ó Tiempo Eficaz (TE). Es el tiempo transcurrido entre el momento que se alcanza la CME y el momento en que el nivel plasmático del fármaco comienza a descender g-Area Bajo la Curva (AUB ó ABC) es una medida de la cantidad del fármaco que llega a la sangre
. Es el tiempo transcurrido entre el momento que se alcanza la CME y el momento en que el nivel plasmático del fármaco comienza a descender. g-Area Bajo la Curva (AUB ó ABC) es una medida de la cantidad del fármaco que llega a la sangre.")
26
La concentración de un fármaco en la biofase varía de un individuo a otro, ó sea, la misma dosis administrada a varios individuos, no produce efectos iguales en todos. Esto de debe a diferencias en la absorción, distribución y eliminación originadas por:

27
-Factores Fisiológicos
-Factores Fisiológicos. Edad, sexo, herencia ó patrón genético, hábitos tóxicos, nutrición, etc. -Factores Patológicos: Enfermedades o procesos patológicos que alteren la función renal, digestiva, cardiovascular, hepática, etc.

28
-Factores yatrogénicos: interacciones de fármacos que puedan alterar sus características farmacocinéticos

29
El conocimiento adecuado de todos estos procesos permite establecer la dosis, la vía de administración y el intervalo de administración más conveniente para lograr una eficacia máxima con el mínimo riesgo para el paciente

30
ABSORCIÓN: Absorción. Es el paso de los fármacos hacia la circulación sistémica y de aquí a los diversos tejidos; esto depende de la fijación a la proteínas plasmáticas, ya que sólo la fracción libre difunde hacia los tejidos. Cualquier desplazamiento de una molécula farmacológica dentro del organismo debe atravesar las membranas biológicas

31
Mecanismos mediante los cuales los fármacos atraviesan las membranas celulares
PROCESOS PASIVOS a-Filtración a través de los poros de la membrana; se considera un proceso de difusión pasiva que involucra la circulación de gran cantidad de agua. Además influyen el tamaño de la molécula y la carga, las moléculas pequeñas y con carga negativa pasarán mejor, ya que la pared de los poros están revestidos de proteínas con carga positiva.

32
Otro elemento a tener en cuenta en este proceso es el tamaño de los poros, muy pequeño en las membranas de los glóbulos rojos, el epitelio intestinal, y la mayoría de las membranas celulares; muy grandes en las membranas celulares de los endotelios vasculares, lo que facilita el acceso al espacio intersticial de los fármacos a través de los capilares sanguíneos

33
b-Difusión Pasiva. Este es el mecanismo más utilizado por los fármacos para atravesar la membrana; la velocidad de difusión depende de varios factores, como son el gradiente de concentración, tamaño y naturaleza de la molécula y de su liposolubilidad

34
La mayoría de los medicamentos son ácidos o bases débiles, y son por lo tanto electrolitos en solución acuosa que se encuentran en forma no ionizada, que atraviesan más fácilmente la membrana, ó en forma ionizada que presentan más dificultad para difundir a través de la membrana.

36
El grado de ionización depende de la naturaleza ácida ó básica del fármaco, del pKa y del pH del medio El pKa se define como el logaritmo negativo de la constante de ionización de una sustancia. Los medicamentos ácidos con un pKa alto son ácidos débiles y se ionizan con dificultad, se absorben mejor en medios ácidos.

37
Los básicos con pKa bajo son bases débiles y se ionizan con dificultad también, y se absorben mejor en medios alcalinos. Por lo tanto las modificaciones del pH del medio controlan el paso de los fármacos a través de las membranas biológicas.

38
Transporte Activo Este tipo de transporte puede ser inhibido por:
-Mecanismos ó sustancias que interfieran en la producción de energía (cambios de temperatura, atmósfera anaeróbica, etc.) -Sustancias que interfieren en las proteínas implicadas en el transporte.
 -Sustancias que interfieren en las proteínas implicadas en el transporte.")
39
-Carencia ó déficit de sustancias necesarias para la síntesis ó funcionamiento de las proteínas transportadoras. Pocos fármacos se absorben activamente, y las áreas donde suele observarse son: túbulo proximal renal, intestino delgado, tracto biliar, hepatocitos, del LCR a la sangre y de la sangre a la saliva

40
OTROS SISTEMAS DE TRANSPORTE.
a-Difusión facilitada. Es un proceso donde no se requiere energía, y se realiza a favor de un gradiente de concentración. Si se requiere de una proteína transportadora de la membrana, la cual se une al sustrato en cuestión, formando un complejo que aumenta la liposolubilidad y favorece el paso a través de la membrana.

41
Este tipo de transporte es utilizado por moléculas demasiado grandes que no pueden atravesar los poros y que por su polaridad tampoco pueden hacerlo. Estos procesos son saturables y específicos

42
b-Endocitosis y Exocitosis
b-Endocitosis y Exocitosis. Son mecanismos mediante los cuales macromoléculas ó partículas pueden entrar ó salir de ellas y conllevan a la rotura de la membrana celular. La endocitosis supone el englobamiento de las partículas mediante la invaginación de la membrana, que al fusionarse posteriormente da origen a una vacuola.

43
La exocitosis es el proceso contrario, en este caso la membrana se abre para permitir la salida de componentes celulares. Para este proceso es necesario la presencia de Ca extracelular

44
c-Utilización de Ionóforos
c-Utilización de Ionóforos. Este tipo de transporte se realiza a través de unas pequeñas moléculas hidrófobas que se disuelven en las bicapas lipídicas y aumentan la permeabilidad de iones específicos. Se distinguen 2 tipos: -Los transportadores móviles de iones (valinomicina) -Los formadores de canales (gramicidina)
 -Los formadores de canales (gramicidina)")
45
d-Utilización de Liposomas
d-Utilización de Liposomas. Los Liposomas son vesículas sintéticas formadas por una ó más bicapas de fosfolípidos que pueden llevar en su interior fármacos hidrosolubles, liposolubles, macromoléculas, material genético, etc.

46
Estos pueden hacer llegar fármacos a diversos tipos celulares de manera específica, logrando la liberación selectiva de ellos; generalmente este tipo de formulación son bien toleradas, pero suelen ser más caras. Son captadas sobre todo por células del SRE

47
e-Transporte a través de hendiduras intercelulares
e-Transporte a través de hendiduras intercelulares. Se observa en la pared de los capilares sanguíneos, en donde los fármacos pasan al espacio intersticial por mecanismos de filtración a través de las hendiduras existentes entre las células

48
FACTORES DE LOS QUE DEPENDE LA ABSORCIÓN DE LOS FÁRMACOS
a-Características fisicoquímicas (peso molecular, carácter ácido ó básico, su pKa, liposolubilidad) b-Características de la preparación farmacéutica (solución oral, polvo, cápsulas, comprimidos, solución IV), además el tamaño de las partículas, presencia de aditivos o excipientes
 b-Características de la preparación farmacéutica (solución oral, polvo, cápsulas, comprimidos, solución IV), además el tamaño de las partículas, presencia de aditivos o excipientes.")
49
c-Características del lugar de absorción; en general será más rápida cuanto mayor y más prolongado sea el contacto con el área de absorción (pH, flujo sanguíneo, motilidad gastrointestinal)
")
50
d-Eliminación presistémica
d-Eliminación presistémica. Por cualquier vía, excepto la intravenosa puede haber una parte del fármaco administrado que no llegue a la circulación sistémica, ya sea porque es eliminado ó por ser destruido previamente. Algunos ejemplos sobretodo para la vía oral son la eliminación por las heces fecales ante de que se complete la absorción, inactivación por el pH del medio ó por las enzimas digestivas.

51
Metabolismo en su paso desde el intestino a través del hígado por el sistema de la vena porta, ó a través del pulmón, antes de llegar a los tejidos donde deben ejercer su efecto. En ambos órganos pero sobre todo en hígado, el fármaco puede sufrir un primer proceso de metabolización denominado FENOMENO DE PRIMER PASO, y por este motivo llegar menos cantidad a la circulación sistémica para ejercer su efecto.

52
FACTORES QUE FAVORECEN LA ABSORCIÓN POR ESTA VÍA
-Grado de ionización -Tiempo de contacto con la mucosa -Capacidad de resistir la actividad del pH gástrico

53
CINETICA DE ABSORCIÓN Estudia la velocidad de absorción, que es la cantidad de fármaco que se absorbe en la unidad de tiempo. Este valor (dc/dt) representa la variación de la concentración en función del tiempo y puede calcularse mediante la ecuación clásica de orden uno. dc/dt = Ka A
 representa la variación de la concentración en función del tiempo y puede calcularse mediante la ecuación clásica de orden uno. dc/dt = Ka A.")
54
La velocidad de absorción depende de Ka que es la constante de velocidad intrínseca del procesos de la absorción. Representa la probabilidad que tiene una molécula de absorberse en la unidad de tiempo, cuanto mayor es Ka, mayor es la velocidad con que se absorbe el fármaco.

55
Se representa en tiempo recíproco; si un fármaco tiene una Ka = 0,05h-1, significa que en una hora se absorbe aproximadamente el 5%. Al principio de la administración del fármaco la velocidad de absorción es mayor y conforme va adsorbiendose esta va disminuyendo.

56
La velocidad de absorción también es directamente proporcional al número de moléculas disponibles que están en solución para absorberse, es decir la concentración remanente de fármaco que aún puede absorberse , a la que llamamos A

57
La semivida de absorción es el tiempo que tarda en reducirse a la mitad el número de moléculas disponibles para absorberse se expresa (t1/2a). Cuanto mayor sea la t1/2a menor será la velocidad de absorción y viceversa

58
TIPOS DE CINETICA DE ABSORCION
Pueden ser de dos tipos: 1-De primer orden; en este caso la velocidad de absorción es proporcional al número de moléculas disponibles para absorberse. Constituye el tipo mediante el cual se realizan la mayoría de los procesos de absorción de los fármacos.

59
2-De orden cero; en este tipo de proceso, el número de moléculas disponibles para absorberse, o que penetran al organismo permanece constante y es independiente de la concentración de moléculas que quedan por absorberse. Se observa en los preparados de liberación prolongada por vía oral, la infusión continua, ó en la administración inhalatoria de gases anestésicos

60
3-Existen procesos activos con una cinética de orden mixto, que se rigen por la ecuación de Michaelis-Menten. Por esta ecuación se rigen todos los procesos saturables, o sea los que pueden incluirse con propiedad dentro de la denominación de procesos bioquímicos

61
BIODISPONIBILIDAD Indica la cantidad y la forma en que un fármaco llega a la circulación sistémica y por tanto está disponible para llegar a los tejidos y producir su efecto. Suele valorarse mediante el área bajo la curva. Únicamente puede medirse en relación con una fórmula de referencia; generalmente se comparan formas orales con una fórmula de referencia inyectada por vía IV

62
DISTRIBUCION Este proceso estudia el transporte del fármaco dentro del compartimiento sanguíneo y su posterior penetración en los tejidos.

63
TRANSPORTE DE LOS FARMACOS EN LA SANGRE
Este se puede realizar por tres formas: -Disueltas en el plasma (fracción libre) -Incorporados en las célula sanguíneas (sobre todo hematíes) -Fijados a las proteínas plasmáticas (sobre todo a la albúmina, que posee mayor superficie y capacidad de fijación)
 -Incorporados en las célula sanguíneas (sobre todo hematíes) -Fijados a las proteínas plasmáticas (sobre todo a la albúmina, que posee mayor superficie y capacidad de fijación)")
64
Se han descrito hasta cuatro sitios diferentes para la unión de fármacos en la albúmina.
Los ácidos débiles se unen casi exclusivamente a la albúmina y pueden hacerlo en los sitios I y II, las bases débiles y los no ionizados liposolubles se unen a las lipoproteínas del plasma, y las bases débiles también pueden hacerlo a la α-glucoproteina.

65
El proceso de unión de los fármacos a las proteínas es reversible y saturable.
Como consecuencia de su unión a las proteínas plasmáticas, los fármacos no producen efectos biológicos, pero esta unión permite su transporte y almacenamiento en el organismo y es uno de los mecanismos más importantes para mantener los niveles plasmáticos y las acciones farmacológicas

66
Sólo el fármaco libre difunde a los tejidos diana y a los órganos de metabolismo y excreción.
El fármaco unido a las proteínas se va liberando poco a poco para alcanzar un equilibrio con la fracción libre en la medida que esta llega a los órganos.

67
El grado de unión a las proteínas plasmáticas es variable
El grado de unión a las proteínas plasmáticas es variable. Algunos fármacos se unen mucho y otros menos. Además existen condiciones ó factores que pueden alterar la unión de los fármacos en base al aumento ó la disminución de la presencia de albúmina plasmática

68
FACTORES QUE DISMINUYEN
-Cirugías -Edad del paciente (RN y ancianos) -Embarazo -Insuficiencia Renal -Quemaduras -Malnutrición grave -Neoplasias -Traumatismos, etc.
 -Embarazo. -Insuficiencia Renal. -Quemaduras. -Malnutrición grave. -Neoplasias. -Traumatismos, etc.")
69
FACTORES QUE AUMENTAN -Ejercicio
-Trastornos psiquiátricos (Neurosis y Psicosis) -Hipotiroidismo -Tumores benignos
 -Hipotiroidismo. -Tumores benignos.")
70
ACCESO A LOS TEJIDOS El paso de los fármacos a los distintos tejidos es muy variable. Básicamente ocurre por difusión pasiva a través de los capilares si son liposolubles, y por filtración si son hidrosolubles.

71
Sólo la fracción libre plasmática difunde hacia el líquido intersticial.
Las características de las membranas endoteliales y la forma de la pared capilar también interviene en el acceso a los tejidos a través de la resistencia al paso del fármaco, esto es significativo en las células hepáticas donde la resistencia es mínima, sin embargo no ocurre así en los capilares del SNC, del ojo, la circulación fetal, etc

72
Otro elemento a tener en cuenta en este aspecto es el flujo sanguíneo regional; también la afinidad de los fármacos por ciertos tejidos (mayor para los adiposos) Por último ciertas condiciones patológicas pueden alterar el patrón normal de distribución de un fármaco, por ejemplo: inflamación, insuficiencia renal, etc.

73
DEPÓSITOS TISULARES Y REDISTRIBUCIÓN
Los principales depósitos de los fármacos son los tejidos, y es común que se acumulen en órganos diferentes de los órganos diana. Pueden hacerlo de manera general en grasa neutra, hígado, huesos, dientes, piel, aparato gastrointestinal

74
BARRERAS Las barreras son estructuras limitantes de los compartimientos: I-BARRERA HEMATOENCEFALICA. En este concepto se abarca la Hematoencefálica y la Hematocefalorraquídea. Esta barrera confiere cierta impermeabilidad al SNC y es un factor de protección frente a los efectos nocivos de las sustancias que ingresan al organismo

75
Los fármacos pueden llegar al SNC por dos vías:
1-Por difusión a través de las paredes de los capilares cerebrales (estas tienen características anatómicas especiales, ya que no tienen poros ni vesículas pinocíticas, sus células están muy unidas, contienen una membrana basal alrededor del endotelio y contienen una capa de pericitos.

76
Los Perocitos son células en forma de araña que se disponen alrededor de los capilares y por último los astrocitos, forman un revestimiento entre los capilares y las células nerviosas que impide también el paso de sustancias)
")
77
Existen estructuras cerebrales que carecen de BHE, ellas son: eminencia media, área postrema en el suelo del IV ventrículo, órgano subfornical, glándula pineal Existen estados patológicos que pueden alterar esta estructura y aumentar la permeabilidad, dentro de estas podemos citar: A-Isquemia

78
B-Lesiones (traumatismos, sustancias citolíticas, etc.)
C-Neoplasias D-Infecciones E-Enfermedades autoinmunes F-Pérdida de los mecanismos de autorregulación (hipertensión endocraneal, estados convulsivos, hipercapnia)
")
79
La velocidad de difusión a través de esta barrera dependerá de la liposolubilidad, del grado de ionización, del pH del plasma, y de la fracción libre de fármaco. Existe un mecanismo de transporte activo, pero este es saturable y con posibilidades de inhibición competitiva.

80
Estas células tienen la capacidad de modificar la estructura de algunos fármacos por mecanismos enzimáticos

81
2-Los fármacos pueden acceder al SNC a través del LCR, formado en los plexos coroideos; los capilares de los plexos coroideos poseen un epitelio formado por células que tienen un borde en cepillo y están acopladas por uniones muy estrechas.

82
Al igual que el caso anterior la velocidad de difusión depende de los mismos factores mencionados, pero aquí se debe tener en cuenta que el LCR de cada espacio tiene características diferentes según el territorio del SNC que este irrigando

83
BARRERA PLACENTARIA Los fármacos administrados a la madre son capaces de atravesar la barrera placentaria y entrar en la circulación fetal, a través del mecanismo de difusión pasiva y cumpliendo sus leyes. Estos pueden afectar al feto a lo largo de toda la gestación y también en el momento del parto.

84
Durante el primer trimestre suelen ser teratogénicos ó sea provocan alteraciones morfológicas, en otras etapas pueden producir alteraciones funcionales del feto, ó tener efectos terapéuticos. El pH de la sangre fetal suele ser un poco mayor que la materna, lo que favorece el acúmulo de fármacos de carácter básico.

85
Dentro del feto, los fármacos se distribuyen de acuerdo a tejidos fetales y etapa de formación.
El hígado fetal y la placenta tiene capacidad metabolizadora, por lo que los efectos en la madre y el feto pueden ser diferentes

86
CINETICA DE DISTRIBUCION
En el organismo los fármacos se encuentran en forma dinámica. Sin embargo en ocasiones es necesario considerar este proceso de distribución de manera estática. Para ello se realizan estudios en modelos compartimentales.

87
Compartimiento: se define como un conjunto de estructuras ó territorios a los que los que un fármaco llega de manera similar y en los cuales se distribuye uniformemente.

88
En la práctica clínica este número de compartimientos se clasifican en:
1-Compartimiento central: tejidos bien irrigados (corazón, pulmón, hígado, riñón, glándulas endocrinas y SNC, en dependencia del paso por la BHE)
")
89
2-Superficial Periférico: tejidos menos irrigados (piel, grasa, médula ósea) y depósitos tisulares a los que el fármaco se une más débilmente
 y depósitos tisulares a los que el fármaco se une más débilmente")
90
3-Periférico profundo: constituido por depósitos tisulares a los que el fármaco se une fuertemente y se libera con mayor lentitud. En base a esto se plantea que los fármacos se pueden distribuir en estas tres variantes de modelos físicos aplicables a los compartimentos

91
MODELO MONOCOMPARTIMENTAL
Aquí se distribuye rápida y uniformemente por todo el organismo. Se suele observar un paralelismo entre las concentraciones plasmáticas y los efectos farmacológicos.

92
MODELO BICOMPARTIMENTAL
Este es el que mejor se ajusta a la mayoría de los fármacos; el fármaco difunde con rapidez al compartimiento central, pero el equilibrio con el periférico se alcanza más lentamente. En estos casos si el efecto farmacológico se produce en el compartimiento central se observa el paralelismo entre las concentraciones plasmáticas y el efecto.

93
Pero si el efecto se lleva a cabo en el periférico, se produce una disociación al inicio, ya que existen altas concentraciones plasmáticas y poco efecto farmacológico; sólo se volverá al paralelismo entre la concentración plasmática y el efecto farmacológico en la fase postdistributiva

94
MODELO TRICOMPARTIMENTAL
En este caso se observa al inicio concentraciones altas en el compartimiento central, y posteriormente una fase de equilibrio en el compartimiento periférico superficial.

95
Sin embargo existe una fase de distribución aún más lenta donde el fármaco se acumula en tejidos específicos con mayor capacidad de retención, que se corresponde con el compartimiento periférico profundo, y si el efecto farmacológico se produce en este compartimiento, el efecto tardará más en aparecer y desaparecerá también más tarde

96
VOLUMEN APARENTE DE DISTRIBUCION
Es un parámetro numérico representativo de la distribución de los fármacos, se obtiene a partir de datos analíticos experimentales. Relaciona la cantidad total del fármaco en el organismo con su concentración plasmática.

97
Este parámetro se utiliza para calcular la dosis inicial que debe administrase para conseguir con rapidez niveles terapéuticos en situaciones de urgencia y es característico de cada fármaco

98
El volumen real en que se distribuyen los fármacos en el organismo depende de las características fisicoquímicas de los mismos, y de la proporción de agua por kilogramo de peso, que varía según la edad, 85% en RN y 65% en los adultos.

99
ELIMINACION CONCEPTO. Proceso mediante el cual una sustancia pasa del medio interno al externo En el caso de los fármacos esto se lleva a cabo a través de: 1-Biotransformación o metabolismo 2-Excreción.

100
BIOTRANSFORMACION O METABOLISMO
Cambios químicos que sufren las sustancias extrañas en el organismo para poder eliminarse mejor. Este proceso generalmente produce inactivación del compuesto original, y produce metabolitos que en el caso de los fármacos algunos son igual ó más activos que ellos

101
Estos metabolitos ejercen efectos iguales ó diferentes a sus moléculas madres y pueden producir efectos tóxicos a veces importantes Los procesos de biotransformación se llevan a cabo en varios órganos, hígado fundamentalmente, en intestino delgado, riñón, sangre, pulmón, placenta, etc.

102
Todos los procesos de biotransformación se llevan a cabo en dos fases ó etapas:
a-Fase I. Aquí se añaden ó se liberan a la molécula, elementos que aumentan su ionización ó su hidrosolubilidad. Las reacciones de esta fase no son sintéticas y producen inactivación, activación ó cambios en la actividad del fármaco. En esta fase se incluyen reacciones de oxidación, reducción e hidrólisis.

103
b-Fase II. En esta fase el compuesto resultante de la fase anterior se une a compuestos endógenos poco liposolubles como ácido glucorónico, acético ó sulfúrico, que aumentan el tamaño de la molécula. Con esto se inactiva el fármaco y aumenta la hidrosolubilidad facilitando su excreción por la orina ó por la bilis. En esta fase II sólo se producen reacciones de síntesis y/ó conjugación.

104
BIOTRANSFORMACION MICROSOMAL
Ocurre a nivel del RE liso de los hepatocitos, participan enzimas oxidativas y es el más utilizado en la biotransformación de los fármacos Las enzimas oxidativas que participan en este proceso utilizan una molécula de O2 para cada molécula del fármaco y sólo toman un átomo de oxígeno para la oxidación del sustrato, el otro átomo se reduce y se forma agua; por eso se llaman oxidasas de función mixta.

105
La oxidasa terminal es una hemoproteina especial llamada citocromo P-450.
Ellas se encuentran en el hígado y en las células de la pared intestinal, también en riñón y mitocondrias de las glándulas suprarrenales. Comprenden una gran familia de enzimas relacionadas, en la especie humana se han identificado de 25 a 30 de ellas.

106
Se nombran con el prefijo CYP seguido de un # que designa la familia, una letra que simboliza la subfamilia y un número que indica el gen productor. Las tres familias involucradas en el metabolismo de los fármacos son la CYP1, CYP2 y CYP3

107
Las enzimas microsomales también catalizan las reacciones de conjugación de glucurónidos. Estas enzimas pueden ser inducidas por fármacos y sustancias químicas ambientales e inhibidos por otras sustancias.

108
FACTORES QUE MODIFICAN LA BIOTRANSFORMACION DE LOS FARMACOS
I-Fisiológicos: -Edad. Prematuros, RN, existe gran inmadurez para los procesos de biotransformación. Ancianos, sobre todo de 65 años en adelante, tienen disminuido el flujo sanguíneo hepático en un 40%, y reducida la función renal

109
-Sexo. Mujer, posee mayor tendencia a efectos tóxicos, por mayor proporción de tejido adiposo y por el tipo de hormonas sexuales. Durante la gestación aumenta la vulnerabilidad por el aumento de los niveles de progesterona, y esta hormona inhibe muchas enzimas de procesos metabólicos.

110
-Herencia. Como consecuencia de una alteración genética, se puede ver alterada la biotransformación de los fármacos (Reacciones Idiosincrásicas) -Dieta. Las proteínas aumentan la actividad hepática microsomal; lo contrario hacen los carbohidratos. El Ca, K y el ácido ascórbico favorecen el metabolismo de los fármacos. Verduras (repollo, coles de Brucelas, rábanos) inducen las enzimas microsomales P-450.
 inducen las enzimas microsomales P-450.")
111
Las metilxantinas (cafeína, teofilina, teobromina) modifican algunos procesos metabólicos
Carnes asadas a la brasa con carbón vegetal aceleran el metabolismo de algunos fármacos por formación de hidrocarburos aromáticos. El jugo de toronja es un inhibidor del CYP3A4 intestinal.

112
II-Farmacológicos: -Inducción enzimática. El suministro de un fármaco puede aumentar la actividad metabólica de la fracción microsomal en varios tejidos. Las consecuencias clínicas de este efecto , puede ser producir un metabolito inactivo (disminución del efecto) ó uno activo (aumento del efecto). Si se administra de forma prolongada un fármaco inductor se puede desarrollar tolerancia farmacológica.
 ó uno activo (aumento del efecto). Si se administra de forma prolongada un fármaco inductor se puede desarrollar tolerancia farmacológica.")
113
Los inductores del citocromo P-450 se clasifican en 5 clases:
a-Tipo fenobarbital. Aumenta la proliferación de RE liso, el peso del hígado, la circulación sanguínea, el flujo biliar, y ciertas proteínas plasmáticas b-Tipo hidrocarburos aromáticos policíclicos. Solo producen un aumento a nivel de la masa hepática sin cambios morfológicos. Se plantea que pueden tener efectos carcinogénicos, por aumento de los productos oxidantes capaces de dañar el ARN

114
c-Tipo esteroides anabolizantes
c-Tipo esteroides anabolizantes. No se conoce el mecanismo a través del cual aumentan el metabolismo microsomal hepático d-Tipo etanol. Actúa como el alcohol, provocando una inducción del metabolismo hepático e-Tipo clofibrato.

115
-Inhibición enzimática
Este proceso puede implicar inhibición competitiva ó no competitiva. Algunas sustancias pueden producir inhibición del sistema microsomal P-450 como insecticidas, monóxido de carbono, sustancias hepatotóxicas, etc.

116
Los fármacos pueden inhibir enzimas que metabolizan sustancias endógenas activas y esto se utiliza en terapéutica, por ejemplo, los fármacos IMAO, utilizados como antidepresivos ó la administración de carbidopa para inhibir la dopa-descarboxilasa en la Enfermedad de Parkinson

117
III-Factores Patológicos.
Alguna enfermedad que altere el metabolismo del fármaco como hepatopatías, insuficiencia renal, trastornos cardiovasculares, etc.

118
VÍAS DE EXCRECION 1-Renal. Es la vía más importante de eliminación de los fármacos, aunque algunos no la utilizan. Algunos fármacos alcanzan concentraciones en orina mucho más elevada que en el plasma. El mecanismo de eliminación renal se realiza utilizando los mismos mecanismos que fisiológicamente forman la orina:

119
-Filtración Glomerular, por los capilares del glomérulo pasan todos los fármacos disueltos en el plasma y con el PM menor de 70,000, no unidos a proteínas plasmáticas

120
-Secreción Tubular. Los fármacos que no pudieron filtrarse, pasan a los túbulos por difusión pasiva ó transporte activo; este puede ser aniones orgánicos (ácidos) ó para cationes orgánicos (bases), ambos sistemas son competitivos y saturables
 ó para cationes orgánicos (bases), ambos sistemas son competitivos y saturables.")
121
-Reabsorción Tubular. El fármaco puede ser reabsorbido por el epitelio tubular y volver a la circulación general. La reabsorción depende del pH de la orina. Como la orina es normalmente ácida, los ácidos débiles son excretados más lentamente, por lo que el metabolismo tiende a transformarlos en ácidos fuertes para eliminarlos más rápido

122
El resultado neto de todos estos procesos es la eliminación de una cantidad de fármaco y sus metabolitos que es cuantificado bajo el concepto de ACLARAMIENTO RENAL Este concepto expresa los ml de plasma ó líquido del cual el riñón elimina totalmente el fármaco en la unidad de tiempo, por lo que se expresa mediante la fórmula

123
FACTORES QUE MODIFICAN LA EXCRECIÓN RENAL
1-Edad 2-El pH de la orina, los ácidos débiles se excretan mejor en orina alcalina, y las bases débiles en orina ácida 3-Diuréticos, estos aumentan el flujo urinario y pueden aumentar la excresión de los medicamentos que se reabsorben 4-Enfermedades renales; aquí se deben evitar fármacos nefrotóxicos, ajustar las dosis, etc.

124
EXCRECION BILIAR Esta vía le sigue en importancia a la renal. Los compuestos que se eliminan por aquí tienen un PM alto; los grupos polares también favorecen la excreción por esta vía, así como fármacos sin capacidad de ionizarse y algunos compuestos organometálicos.

125
Los fármacos pasan de la circulación a la bilis por difusión pasiva ó por transporte activo
Algunos pueden ser reabsorbidos por el intestino y volver a sufrir procesos de biotransformación ó de eliminación renal

126
OTRAS VÍAS DE EXCRECION
La vía pulmonar es importante para los gases y fármacos volátiles. La excreción por el estómago, el intestino delgado y colon, se realiza a través de los transportes de membranas; algunos pueden reabsorberse de nuevo y excretarse de manera más lenta.

127
La excreción por saliva, sudor y las lágrimas es menos importante; por esta vía la eliminación depende de la liposolubilidad del fármaco ó sus metabolitos, para poder atravesar las membranas celulares de estas glándulas

128
En el caso de la saliva, como los fármacos pueden pasar a esta por difusión pasiva, su concentración es similar a la concentración libre en el plasma y esto se utiliza para monitorear las concentraciones libres de algunos fármacos

129
La excreción por la leche materna se produce a través de un mecanismo de paso a través de las membranas celulares de las glándulas, por lo tanto se elimina por esta vía las sustancias liposolubles y la fracción no ionizada de los ácidos y bases débiles.

130
El pH de la leche materna es ligeramente inferior al del plasma por lo que las bases débiles se eliminan mejor. No obstante son eliminados pequeñas cantidades y sólo es importante esta vía para los medicamentos que se consumen en grandes cantidades ó de manera crónica

131
Se consideran peligrosos los fármacos antitiroideos, yodo radiactivo, litio, cloranfenicol, quimioterápicos. Deben vigilarse la administración crónica de isoniacida, sulfas, difenilhidantoina, cafeína, teofilina, barbitúricos, antipalúdicos y salicilatos

132
Otras vías de escasa importancia son la piel y el pelo; son útiles para detectar algunos metales como arsénico y mercurio

133
CINETICA DE ELIMINACION
La cinética de eliminación es parecida a la de absorción. Mide la velocidad de eliminación de los fármacos del organismo y se expresa por una Ke. Esta constante indica la probabilidad de una molécula de eliminarse en la unidad de tiempo, se expresa el tiempo invertido.

134
Por ejemplo Ke = 0,003 h-1, significa que en una hora se elimina el 3%
Estos mecanismos de eliminación pueden ser de:

135
Orden 1: la velocidad de eliminación es mayor, mientras mayor es la concentración plasmática del fármaco. Constituyen la mayoría de los mecanismos de eliminación. Orden 0: esta ocurre cuando se satura el mecanismo de eliminación y el número de moléculas que se elimina permanece constante.

136
La semivida de eliminación t1/2e es el tiempo que tarda un fármaco en reducirse a la mitad su concentración plasmática. Este valor tiene importancia para decidir la pauta de administración.

137
En el modelo monocompartimental el descenso de las concentraciones plasmáticas depende únicamente de la K de eliminación del fármaco. En el compartimiento bicompatimental, la velocidad con que disminuyen las concentraciones plasmáticas depende de la K de eliminación y de los procesos de distribución.

138
CINETICA LINEAL Y NO LINEAL
Decimos que la cinética de un fármaco es lineal, cuando sus constantes de absorción, distribución y eliminación no varían con el tiempo, ni tampoco cuando se modifican las dosis.

139
Por el contrario se dice que un fármaco tiene cinética no lineal, cuando se afectan los procesos de absorción, distribución ó eliminación, pero generalmente se pueden deber a saturación de la unión con proteínas, metabolismo hepático ó transporte renal activo y generalmente afecta el proceso de eliminación

Presentaciones similares










>")


 Lo que el cuerpo le hace al fármaco Terminología Droga: Mezcla de.>")






