Descargar la presentación
La descarga está en progreso. Por favor, espere

1
Errores innatos del metabolismo y otros trastornos genéticos
Cap. 11 Robbins & Cotran Pathology Dr. P Goyenaga

2
ERRORES INNATOS DEL METABOLISMO Y OTROS TRASTORNOS GENÉTICOS
SIR ARCHIBALD BARROD 1908, acuñó el nombre, el número de ha aumentado en forma exponencial La mayoría son raros, se heredan habitualmente como enfermedades autosómicas recesivas o ligadas al cromosoma X, Enfermedades mitocondriales Se citan algunos signos clínicos que sugieren su existencia Tab.10-6, de base en el neonato Se han seleccionado tres defectos genéticos metabólicos para se comentados aquí porque su diagnóstico precoz mediante los programas de cribado neonatal es importante porque una dieta adecuada puede evitar la muerte precoz y complicaciones posteriores. Fenilcetonuria (FCU) , Galactosemia y Fibrosis Quística El cribado neonatal de FQ, sigue siendo tema discutido. Se incluye porque es de las mas frecuentes y potencialmente letal.
 , Galactosemia y Fibrosis Quística. El cribado neonatal de FQ, sigue siendo tema discutido. Se incluye porque es de las mas frecuentes y potencialmente letal.")
3
Anormalidades Sugestivas de errores innatos del metabolismo y otros trastornos genéticos
General Signos dismórficos Sordera Automutilación Pelo Anormal Olor corporal o de la orina (“pies sudados"; “pálidos, húmedos y mohoso"; “jarabe de arce") Hepatoesplenomegalia; cardiomegalia Hidrops
 Hepatoesplenomegalia; cardiomegalia. Hidrops.")
4
Neurológicas Músculo articulaciones
Hipotonía o Hipertonía Miopatía Coma Movimientos anormales Letárgica persistente Convulsiones Gastrointestinales Mala alimentación Vómitos recurrentes Ictericia Ojos Catarata Mácula rojo cereza Luxación del cristalino Glaucoma

5
FENILCETONURIA

6
FENILCETONURIA LOS HOMOCIGOTOS CON FCU – AUTOSOMIC A RECESVA TIENEN UNA DEFICIENCIA GRAVE DE FENIULALANINA HIDROXILASA , LO QUE DA LUGAR A HIPERFENILALANINEMIA Y SUS CONSECUENCIAS GRAVES ESCANDINAVOS. POCO EN AFROAMERICANOS Y JUDIOS R. N. NLS. AL NACER . POCAS SEMANAS , FENILALANINA SERICA AUMENTA M. DAÑO CEREBRAL, < 4% NIÑOS NO TRATADOS TIENEN VALORES I.Q. SUPERIIORES A 50 O 60, DEF. CAMINAR, HABLA, CONVULSIONES, PELO DESPIGMENTADO, PIEL, ECCEMA. MUJERES CON FCU CLINICAMENTE NLS., ABANDONAN TRATAMIENTO EN EDAD FERTIL Y TIENEN HIPERFENILANINEMIA , SE EMBARAZAN = % TIENEN HIJOS CON RETRAZO MENTAL . 15% CARDIOPATIAS INCLUSO AUNQUJE LOS HIJOS SEAN HETEROCIGOTOS. SINDROME FCU MATERNA EFECTOS TERATOGÉNICOS CRUZAN BARRERA PLACENTA CONTROL ANTES DEL EMBARAZO Y DURANTE.

7
FENILCETONURIA LA ANORMALIDAD BIOQUÍMICA FCU ES INCAPACIDAD DE CONVERTIR LA F.A. EN TIROSINA FIG.10-17 EN NIÑOS NLS. ES NECESARIA PARA LA SINTESIS PROTEICA MENOS DEL 50% DE LA INGESTA DE F.A. EL RESTO SE CONVIERTE DE FORMA IRREVERSIBLE EN TIROSINA ATRAVES DEL COMPLEJO ¨SISTEMA FENILALANINA HIDROXILASA HEPÁTICA¨ PAH TIENE OTROS DOS COMPONENTES . EL COFACTOR ¨TETRAHIDROBIOPTERINA¨ BH4 Y LA ENZIMA ¨DIHIDRO PTERIDINA REDUCTASA¨ DHPR QUE REGENERA LA BH4 AUNQUE LA HIPERFENILALANINEMIA NEONATAL PUEDE ESTAR CAUSADA POR DEFICIENCIAS DE ESTOS DOS COMPANENTE , EL 98% DE LOS CASOS SE ATRIBUYE A ANOMALIAS DE ¨FENILALANINA HIDROXILASA PAH¨

8
Figure 10-17 El sistema fenilalanina hidroxilasa
TIENE OTROS DOS COMPONENTES . EL COFACTOR ¨TETRAHIDROBIOPTERINA¨ BH4 Y LA ENZIMA ¨DIHIDRO PTERIDINA REDUCTASA¨ DHPR QUE REGENERA LA BH4

9
Es una rara afección en la cual un bebé nace sin la capacidad para descomponer apropiadamente un aminoácido llamado fenilalanina. Causas, incidencia y factores de riesgo La Fenilcetonuria es una enfermedad hereditaria, lo cual significa que se transmite de padres a hijos. Ambos padres deben transmitir el gen defectuoso para que el bebé padezca la enfermedad, lo que se denomina un rasgo autosómico recesivo. Los bebés con Fenilcetonuria carecen de una enzima denominada fenilalanina hidroxilasa, necesaria para descomponer un aminoácido esencial, llamado fenilalanina, que se encuentra en alimentos que contienen proteína. Sin la enzima, los niveles de fenilalanina y dos substancias estrechamente relacionadas se acumulan en el cuerpo. Estas sustancias son dañinas para el sistema nervioso central y ocasionan daño cerebral.

10
La fenilalanina juega un papel en la producción corporal de melanina, el pigmento responsable del color de la piel y del cabello. Por lo tanto, los niños con esta afección usualmente tienen un cutis, cabello y ojos más claros que sus hermanos o hermanas sin la enfermedad.

11
Retraso de las habilidades mentales y sociales
Tamaño de la cabeza considerablemente por debajo de lo normal Hiperactividad Movimientos espasmódicos de brazos y piernas Retardo mental Convulsiones Erupción cutánea Temblores Postura inusual de las manos Si la afección se deja sin tratamiento o si no se evitan los alimentos que contienen fenilalanina, se puede detectar un olor "a ratón" o "a moho" en el aliento, la piel y la orina. Este olor inusual se debe a la acumulación de sustancias de fenilalanina en el cuerpo

12
Se somete a los recién nacidos a extracciones rutinarias de sangre, que se obtiene al puncionar el talón y se recolecta en un papel secante especial. Posteriormente se realizan exámenes de sangre de rutina, como el de Fenilcetonuria y la determinación del tipo sanguíneo. En muchos hospitales se realizan otros exámenes, tales como el de función tiroidea, el de hemoglobina S (enfermedad drepanocítica) y para el diagnóstico de otros trastornos de la sangre (hemoglobinopatías). Puede realizarse los exámenes de acuerdo a la población, tomando en cuenta la raza y la etnicidad de los recién nacidos para determinar cuáles exámenes de rutina son necesarios
 y para el diagnóstico de otros trastornos de la sangre (hemoglobinopatías). Puede realizarse los exámenes de acuerdo a la población, tomando en cuenta la raza y la etnicidad de los recién nacidos para determinar cuáles exámenes de rutina son necesarios.")
13
La Fenilcetonuria se puede detectar fácilmente con un simple examen de sangre. HNN .
La mayoría de los estados de los Estados Unidos exige una prueba de detección de esta enfermedad para todos los recién nacidos, la cual generalmente se lleva a cabo con una punción en el talón poco después del nacimiento. Si la prueba de detección inicial es positiva, se requieren exámenes posteriores de sangre y orina para confirmar el diagnóstico.

14
La sangre se extrae rutinariamente de un recién nacido para examinarla.
La sangre se obtiene por “punción en el talón” y se recoge en un papel secante especial. Las pruebas de rutina usualmente abarcan: Fenilcetonuria, función tiroidea, hemoglobina S (enfermedad drepanocítica) y se pueden evaluar otros trastornos. Los programas de tamizaje en recién nacidos varían de un estado a otro. Los exámenes se pueden adaptar a la población local, determinando las pruebas de rutina que se deban hacer.
 y se pueden evaluar otros. trastornos. Los programas de tamizaje en recién nacidos varían de un estado a otro. Los exámenes se pueden adaptar a la población local, determinando las pruebas de rutina que se deban hacer.")
15
La fenilalanina se encuentra en cantidades significativas en alimentos como la leche, los huevos y otros alimentos comunes. Además, se encuentra en el edulcorante artificial Nutrasweet (aspartamo), razón por la cual cualquier producto que contenga aspartamo se debe evitar Si este trastorno no recibe tratamiento, se presenta retardo mental severo. El trastorno de hiperactividad y déficit de atención (ADHD, por sus siglas en inglés) parece ser el problema más común que se observa en quienes no siguen estrictamente una dieta muy baja en fenilalanina
 parece ser el problema más común que se observa en quienes no siguen estrictamente una dieta muy baja en fenilalanina.")
16
GALACTOSEMIA

17
GALACTOSEMIA GALACTOSEMIA , AJTOSOMICA RECESIVA DEL METABOLISMO DE LA GALACTOSA. LA LACTOSA , HIDRATO DE CARBONO DE LALECHE DE MAMIFEROS SE DESCOMPONE EN GLUCOSA Y GALACTOSA EN EL INTESTINO POR ACCION DE LACTASA , DESPUES LA GALACOSA SE CONVIERTE EN GLUCOSA EN TRES ETAPAS. DOS VARIANTES DE GALACTOSEMIA 1) FALTA TOTAL DE FALACTOSA 1 FOSFATO URIDIL TRANSFERASA, IMPLICADA EN LA 2a DEFICIENCIA DE ¨GALACTOCINASA¨ (1) QUE DA FORMA LEVE SIN RETRASO MENTAL COMO RESULTADO DE LA DEF. TRANSFERASA SE ACUMULA GALACTOS 1 FOSFATO EN MUCHOS LUGARES, INCLYUENDO BAZO HIGADO (COMO CIRROSIS) CRISTALINO RIÑONES, MUSC. CARDIACO SE ACTIVAN VIAS METABOLICAS ALTERNATIVAS, QUE PRODUCEN GALACTITOL QUE SE ACUMULA EN TEJIDOS. EL CUADRO ES VARIABLE LO QUE REFLEJA LA HETEROGENEIDAD DE LAS MUTACIONES EN EL GEN DE LA ¨GALACTASA-1-FOSFATO-URIDIL TRANSFERASA (GALT) QUE PRODUCE LA GALACTOSEMIA CON AFECCIONES MULTIORGÁNICAS
 FALTA TOTAL DE FALACTOSA 1 FOSFATO URIDIL TRANSFERASA, IMPLICADA EN LA 2a DEFICIENCIA DE ¨GALACTOCINASA¨ (1) QUE DA FORMA LEVE SIN RETRASO MENTAL. COMO RESULTADO DE LA DEF. TRANSFERASA SE ACUMULA GALACTOS 1 FOSFATO EN MUCHOS LUGARES, INCLYUENDO BAZO HIGADO (COMO CIRROSIS) CRISTALINO RIÑONES, MUSC. CARDIACO. SE ACTIVAN VIAS METABOLICAS ALTERNATIVAS, QUE PRODUCEN GALACTITOL QUE SE ACUMULA EN TEJIDOS. EL CUADRO ES VARIABLE LO QUE REFLEJA LA HETEROGENEIDAD DE LAS MUTACIONES EN EL GEN DE LA ¨GALACTASA-1-FOSFATO-URIDIL TRANSFERASA (GALT) QUE PRODUCE LA GALACTOSEMIA CON AFECCIONES MULTIORGÁNICAS.")
18
GALACTOSEMIA HEPATOMEGALIA – CAMBIO GRASO – CICATRIZ – CIRROSIS ALCOHOL SIMILAR. OPACIFICIDAD CRISTALINO (CATARATA) , ABSORVE AGUA Y SE HINCHA SNC. APARECEN ALTERACIONES INESPECÍFICAS INCLUYENDO PÉRDIDA DE CPELULAS DE LOS NERVIOS, GLIOSIS, Y EDEMA, SOBRETODO NÚCLEOS DENTADOS DEL CEREBELO Y LOS NÚCLEOS OLIVARES DE LA MEDULA CASI DESDE EL NACIMIENTO ESTOS RN NO CRECEN . AL CABO DE POCOS DÍAS DE INGERIR LECHE, APARECEN VÓMITOS Y DIARREA, ICTERICIA FISIOLOGICA APARENTA PROLONGARSE. CATARATA A LAS POCAS SEMANAS Y MESES SE DETECTA RETRAZO MENTAL FRECUENCIA AUMENTADA DE ¨SEPTICEMIA POR ESCHERICHIA COLI¨ QUIZA POR DISMINUCIÓN DE ACTIVIDAD BACTERICIDA DE PMN , HEMÓLISIS COAGULOPATÍA PRENATAL POSIBLE DX ANTENATAL POR PRUEBA ACTIVIDAD GALT LIQ., AMNIÓTICO + DE 140 MUTACIONES GALT, + BLANCOS NO HISPANOS. MUCHOS DE LOS CAMBIOS CLÍNICOS Y MORFOLOGICOS DE LA GALCTOSEMIA SE PUEDEN PREVENIR O MEJORAR CON LA RETIRADA DE LA GALACTOSA DE LA DIETA DURANTE LOS PRIMEROS 2 A. DE VIDA. CONTROL DESPUES DEL NACIMIENTO PREVIENE CATARATA Y LESIÓN HEPATICA Y DESARROLLA PRACTICAMENTE NORMAL. PACIENTES MAYORES SUELEN AFECTARSE EN EL HABLA Y FALLO GONADAL , ESP., OVARICO

19
Figure 10-19 Galactosemia. El hígado muestra un cambio graso extenso y una delicada fibrosis

20
Figure 10-19 Galactosemia. El hígado muestra un cambio graso extenso y una delicada fibrosis

21
Figure 10-18 Vías del metabolismo de la galactosa.
Se han identificado dos variantes de Galactosemia. En la forma mas frecuente, hay una falta total de galactosa -1fosfato uridil transferasa implicada en la reacción 2. la variante rara se debe a una deficiencia de galactocinasa, implicada en la reacción 1. Def. Galactocinasa enf. Leve sin retraso mental. Def. de transferasa, se acumula galactosa-1-fosfato en muchos lugares, hígado bazo cristalino riñones musc. Cardiaco corteza cerebral y eritrocitos

22
GALACTOSEMIA puede manifestar ictericia, vómitos, letargo, irritabilidad y convulsiones

23
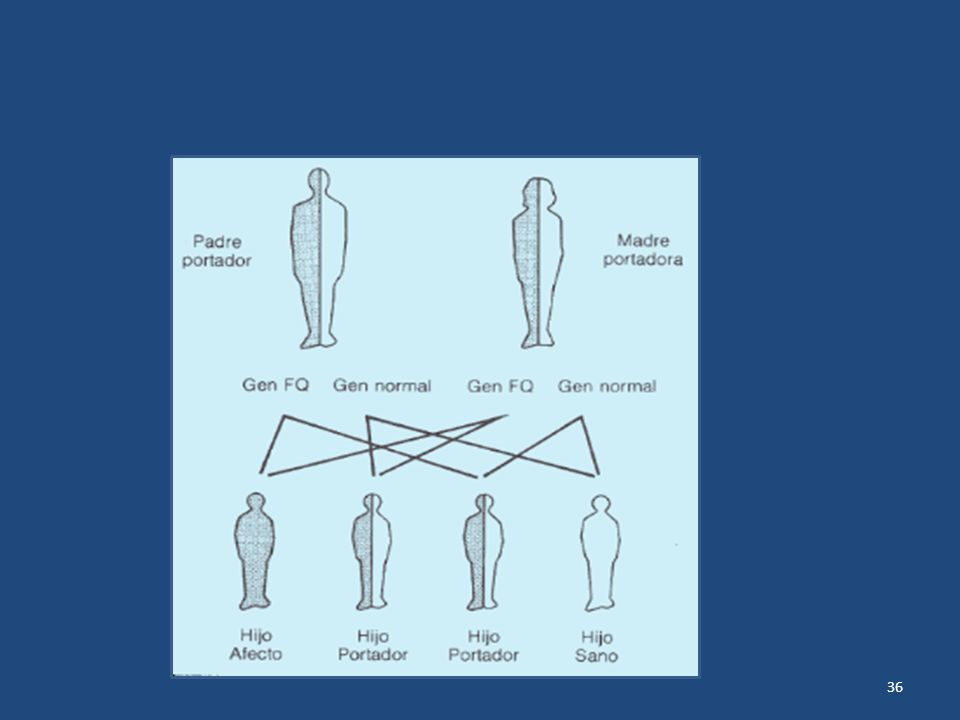
Para padecer esta enfermedad es necesario que el gen que la provoca esté en las células en "doble dosis" (es decir, que el niño haya heredado un gen patológico del padre y otro gen patológico de la madre). Ejemplo: Para que el niño tenga los ojos azules es necesario que ambos genes, el procedente de la madre y el del padre, indiquen que dicho color sea azul. Si uno de los genes heredados no representa el color azul, el niño no tendrá los ojos de ese color. Fig 2.POSIBILIDADES SANO (DOS GENES SANOS ) ENFERMO (DOS GENES ANOMALOS ) PORTADOR SANO (UN GEN ANÓMALO Y OTRO SANO Si los padres no padecen esta enfermedad se debe a que presentan un solo gen anómalo. A este tipo de transmisión se le denomina autosómica recesiva.
 ENFERMO (DOS GENES ANOMALOS ) PORTADOR SANO (UN GEN ANÓMALO Y OTRO SANO. Si los padres no padecen esta enfermedad se debe a que presentan un solo gen anómalo. A este tipo de transmisión se le denomina autosómica recesiva.")
24
La galactosemia es una enfermedad genética que genera una deficiencia enzimática y se manifiesta
con incapacidad de utilizar el azúcar simple galactosa, lo cual provoca una acumulación de ésta dentro del organismo, produciendo lesiones en el hígado y el sistema nervioso central. Galactosemia Galactosa La Galactosemia es una enfermedad autosómica recesiva caracterizada por la incapacidad de convertir la galactosa en glucosa. Se reconocen dos procesos, la galactosemia clásica, que se debe a la deficiencia de la enzima galactosa-1-fosfatouridil-transferasa y se asocia con la formación de cataratas, retardo mental y cirrosis. El segundo tipo de galactosemia se produce por deficiencia de la enzima galactoquinasa, y solo se manifiesta con cataratas.

25
FIBROSIS QUISTICA (MUCOVISCIDOSIS)
")
26
FIBROSIS QUISTICA - MUCOVISIDOSIS
Los niños del beso salado "The History of Cystic Fibrosis“ Desde la edad media se decía: Aquel niño que al besarlo sabe salado, esta embrujado y no tardará en morir. FIBROSIS QUISTICA - MUCOVISIDOSIS TRASTORNO DIFUSO EN EL TRANSPORTE EPITELIALQUE AFECTA A LA SECRECIÓN DE LÍQUIDO EN LAS GLÁNDULAS EXOCRINAS Y LA CAPA EPITELIAL DE LOS TRACTOS RESPIRATORIOS GASTROINTESTINAL Y REPRODUCTOR

27
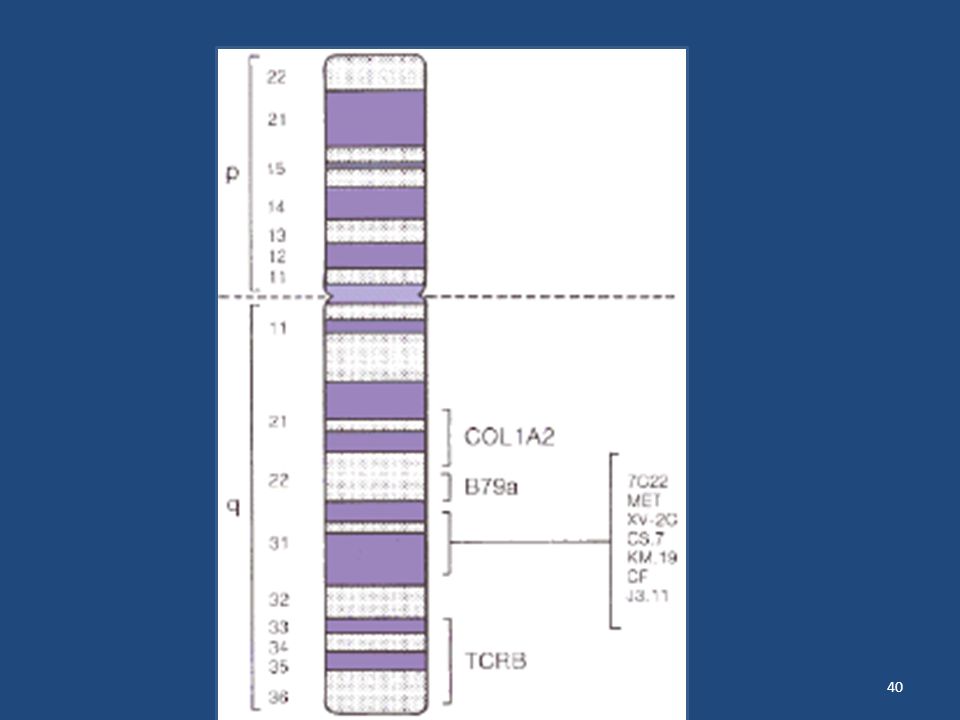
La fibrosis quística o mucovicidosis del páncreas es la enfermedad hereditaria autosómica y recesiva mas frecuente en las poblaciones de raza blanca (*uno por cada 2000 nacidos vivos). Se caracteriza por la producción de un mucus espeso, insuficiencia pancreática exógena y electrolitos altos en el sudor. En ausencia de tratamiento es mortal y con tratamiento rara vez superan la adolescencia. La causa es un defecto en un único gen que se encuentra en el brazo largo del cromosoma 7.

28
Activación del regulador normal de la conductancia transmembrana(CFTR) en la fibrosis quística CFTR normal. El CFTR consta de dos dominios transmembrana, dos dominios de unión con nucleótidos (NBD) y un dominio regulador R . Los antagonistas p.ej. Acetilcolina se unen a las células epiteliales e incrementan el AMPc lo que activa la proteín cinasa A, que fosforila el CFTR en el dominio R lo que da lugar a la abertura del canal de cloro. ABAJO , CFTR desde el gen hasta la proteína. La mutación mas frecuente en el gen del CFTR da lugar a un pliegue defectuoso de la proteína en el sistema Golgi/RE y a la degradación del CFTR antes de que alcance la superficie celular . Otras mutaciones afectan a la síntesis de CFTR , la unión de los nucleótidos y los dominio R, y los dominios de membrana. CL-

29
El defecto genético, provoca que una proteína denominada CFTR se cree alterada en todas las células. Esta proteína se ubica en la pared celular, en los denominados "Canales del Cloro" por donde pasan los iones del interior de la célula al exterior y viceversa. Al no funcionar bien esta proteína, por distintos motivos, los iones de cloro no pasan correctamente, o pasan en poca cantidad, por estos canales. El cloro arrastra consigo agua y otros iones. El resultado final es que las segregaciones de todas las glándulas exocrinas son anormalmente espesas en las personas afectadas de FQ

30
Fig Defectos en el Canal de Cloro en conducto sudoríparo (arriba) causan incrementos de cloro y sodio en el sudor. En la vía respiratoria (abajo), los pacientes con F.Q. presentan disminución en la secreción de cloro e incremento en la reabsorción sodio y agua con deshidratación de la cubierta mucosa de las células epiteliales , alteraciones en la acción mucociliar y tapones mucosos en las vías respiratorias. CFTR, los pacientes tienen una actividad antibacteriana deficiente.
 causan incrementos de cloro y sodio en el sudor. En la vía respiratoria (abajo), los pacientes con F.Q. presentan disminución en la secreción de cloro e incremento en la reabsorción sodio y agua con deshidratación de la cubierta mucosa de las células epiteliales , alteraciones en la acción mucociliar y tapones mucosos en las vías respiratorias. CFTR, los pacientes tienen una actividad antibacteriana deficiente..")
31
Las muchas manifestaciones clínicas de las mutaciones en el gen de la fibrosis quística , desde la mas grave hasta la asintomática

32
La Fibrosis Quística o Mucovicidosis es la enfermedad genética e incurable más frecuente en la raza blanca. Uno de cada 2500 nacidos está afectado de Fibrosis Quística y una de cada 20 personas es portadora del gen defectuoso de la FQ y la puede trasmitir a sus hijos. Cuando en una pareja los dos son portadores de este gen, existe un 25% de probabilidad de que cada hijo sea enfermo de Fibrosis Quística. Es una enfermedad multisistémica, es decir se manifiesta en diferentes sistemas y aparatos del cuerpo humano.

33
La Fibrosis Quística es una enfermedad autosómica recesiva.
No se encuentra en los cromosomas sexuales y precisa de ambos genes para que se pueda manifestar el desarrollo de la misma. En el caso de la Fibrosis Quística se manifiesta cuando se han heredado dos genes alterados, manifestando en consecuencia el enfermo fibroquístico, los síntomas normales de la enfermedad.

34
La fibrosis quística es la causa más común de la enfermedad pulmonar crónica en los niños y jóvenes y es el trastorno hereditario mortal más común que afecta a los caucásicos

35
La malformación de los dedos es síntoma de una enfermedad, por lo general, del corazón o de los pulmones, lo cual produce niveles sanguíneos crónicamente bajos. Las enfermedades que causan malabsorción, como la fibrosis quística o la enfermedad celíaca

37
Las personas que tienen un gen normal y un gen enfermo, son portadores sanos de la enfermedad y no manifiesta ninguno de los síntomas comunes a la misma. Cuando dos padres portadores tienen hijos, existen unas posibilidades de que nazcan hijos sanos, portadores sanos y afectos: - En el caso de que ambos progenitores sean portadores sanos las probabilidades son que un 25 % de los hijos sean totalmente sanos, un 50% sean portadores sanos y un 25 %, sean afectados de Fibrosis Quística. - En el caso de que un portador sano engendre hijos con una persona totalmente sana, la posibilidad de que nazcan afectados F.Q. no existe pero sí de que nazcan más portadores sanos.

38
El GEN de la F.Q. Las investigaciones en el campo de la genética de la F.Q. han permitido realizar avances muy importantes en los últimos años. El gen de la F.Q. pudo ser localizado en el cromosoma 7. Existen varios marcadores del ADN que detectan variaciones, que sirven para seguir la herencia del gen F.Q. en el seno de las distintas familias. Estos marcadores permiten el diagnóstico prenatal y saber si los otros hijos de la pareja son portadores de la enfermedad.

39
La identificación del gen F. Q
La identificación del gen F.Q. ha supuesto un considerable avance en el estudio de la enfermedad. Tras las investigaciones realizadas en Gran Bretaña, España, Canadá y Estados Unidos, los científicos de estos dos últimos países identificaron, a mediados de 1989, el gen responsable de la enfermedad. Se trata un gen muy grande que tiene unas bases o nucleótidos. Un error en una sola de estas unidades puede ser fatal y producir la enfermedad. En la mayoría de los casos se ha podido ver que el defecto es debido a la pérdida de tres bases, dando lugar a la ausencia de un aminoácido denominado fenilalanina en la posición 508. Esta mutación o defecto, ha sido encontrada en el 75 % de los cromosomas F.Q.

41
Los efectos más destacables

42
La expectativa de vida promedio para las personas que tienen FQ es de 33 años. La FQ no afecta a la inteligencia Muchos de estos niños también sufren diabetes por culpa de la FQ

43
Pulmones de un paciente fallecido por fibrosis quistica
Pulmones de un paciente fallecido por fibrosis quistica. Se observa abundantes tapones de moco y dilatación del árbol traqueobronquial . Parénquima pulmonar consolidado por combinación de secreciones y neumonía Color verde asociado a infecciones por Pseudomonas

44
Alteraciones de la fibrosis quística de grado leve - moderado en el páncreas aparecen dilatados ocupados por material mucinoso eosinófilo y las glándulas del parénquima están atróficas y sustituidas por tejido fibroso

45
1. Enfermedad senopulmonar crónica manifestada por.
Tabla 10-7 Signos clínicos y criterios diagnósticos de fibrosis quística 1. Enfermedad senopulmonar crónica manifestada por. A. Colonización /infección persistente con microorganismos típicos de F.Q. Stataphylococcus aureus, Haemophilus inf. Pseudomonas auruginosa mucoide y no mucoide , Burkholderia cepacia B. Tos y producción de esputo crónico C. Anomalías persistentes en la Rx de Tx.(bronquiectasias, atelectasias , infiltrados hiperinsuflación D. Obstrucc-. De vías aéreas manifestada como sibilancias y atrapamiento aéreo E. Pólipos nasales, anormales de los senos paranasales en la Rx o la TAC F. Acropaquia. 2. Anomalías gastrointestinales y nutricionales , incluyendo. Intestinales, incluyendo ilio meconial, síndrome de obst. intest. Distal prolapso rectal Pancreáticas, insuficiencia pancrea., pancreatitis recurrente Hepáticas, enf. Hepática cro. Manifestada como la evidencia clínica o histológica de cirrosis biliar focal, o cirrosis multilobular Nutricionales , retraso de crecimiento (malnutrición proteico calórica) Hipoproteinemia , edema, complicaciones secundarias a la defic. De vit. Liposolubles.
 Hipoproteinemia , edema, complicaciones secundarias a la defic. De vit. Liposolubles.")
46
Signos clínicos y criterios diagnósticos de F.Q.
3. Síndrome de pérdida salina depleción aguda de sal, acidosis metabólica crónica 4. Anomalías urogenitales del varón que dan lugar a azoospermia obstructiva (ausencia bilateral congénita del conducto deferente) CRITERIOS DIAGNÓSTICOS DE FIBROSIS QUISTICA Uno o más signos fenotípicos característicos , 0 una historia de fibrosis quística en un hermano 0 un resultado positivo en una prueba de cribado neonatal Y Un aumento de la concentración de cloro en el sudor en una o más determinaciones 0 identificación de dos mutaciones de fibrosis quística 0 demostración de transporte iónico epitelial nasal anormal.
 CRITERIOS DIAGNÓSTICOS DE FIBROSIS QUISTICA. Uno o más signos fenotípicos característicos , 0 una historia de fibrosis quística en un hermano. 0 un resultado positivo en una prueba de cribado neonatal. Y. Un aumento de la concentración de cloro en el sudor en una o más determinaciones. 0 identificación de dos mutaciones de fibrosis quística. 0 demostración de transporte iónico epitelial nasal anormal.")
47
Existen de 6 a 12 posiciones que una persona con enfermedad pulmonar puede adoptar para drenar el moco desde ciertas partes de los pulmones. Otra persona puede golpear ligeramente en ciertas áreas para ayudar a aflojar el moco y permitir que éste sea expulsado. Otras formas de aliviar la congestión pulmonar por fibrosis quística o bronquiectasia son los aerosoles

48
El gen responsable de la FQ fue clonado en 1989 y codifica para la proteína denominada reguladora de conductancia transmembrana de la FQ (CFTR). Este gen está ubicado en el brazo largo del cromosoma 7, posee 27 exones, un tamaño aproximado de 230 kilobases (Kb) y produce un ARN mensajero de 6,5 kb. El producto normal del gen es una proteína de membrana de 1 480 aminoácidos, que funciona como canal de cloruro regulado por AMP cíclico y que se expresa casi exclusivamente en las células de los epitelios secretores. Su estructura comprende dos dominios transmembrana (TM), dos regiones citoplasmáticas que unen ATP (NBD) y un dominio con residuos susceptibles a fosforilación por la proteína quinasa dependiente de AMPc (R). Los dominios transmembrana contribuyen a la formación del poro del canal, en tanto los dominios NBD y R regulan su actividad1. La FQ es una condición de herencia autosómica recesiva. Esto implica que ambos padres de un niño con esta enfermedad son portadores (aunque existen excepciones, como la disomía uniparental) y, por lo tanto, tienen un riesgo de recurrencia de 25% para cada embarazo, y que cada hijo sano tiene 2/3 de probabilidades de ser portador. Desde un punto de vista poblacional, dada una frecuencia de la enfermedad de 1/4000 a 1/4500, 1 cada 32 personas sanas serían portadoras2
, dos regiones citoplasmáticas que unen ATP (NBD) y un dominio con residuos susceptibles a fosforilación por la proteína quinasa dependiente de AMPc (R). Los dominios transmembrana contribuyen a la formación del poro del canal, en tanto los dominios NBD y R regulan su actividad1. La FQ es una condición de herencia autosómica recesiva. Esto implica que ambos padres de un niño con esta enfermedad son portadores (aunque existen excepciones, como la disomía uniparental) y, por lo tanto, tienen un riesgo de recurrencia de 25% para cada embarazo, y que cada hijo sano tiene 2/3 de probabilidades de ser portador. Desde un punto de vista poblacional, dada una frecuencia de la enfermedad de 1/4000 a 1/4500, 1 cada 32 personas sanas serían portadoras2.")
49
QUIENES TRANSMITEN LA ENFERMEDAD
QUIENES TRANSMITEN LA ENFERMEDAD?: Es imprescindible para que un niño padezca ésta enfermedad que ambos padres le hayan transmitido el gen de la enfermedad. El niño hereda un gen patológico del padre y otro gen patológico de la madre. Es decir, en "dosis doble", los padres sanos del niño enfermo solo tienen un dosis única. ¿Cuál es el riego de que con ambos padres portadores sanos, tenga un hijo FQ?. Un 25 %, que salga un niño con FQ, es decir 1 de cuatro. Un 50 %, que salga un niño portador sano Un 25 %, que salga un niño que ni siquiera sea portador sano. ¿CON QUE FRECUENCIA SE PRODUCE?: Raza Blanca - 1 de cada nacimientos Raza Negra - 1 de cada nacimientos Raza Amarilla - 1 de cada nacimientos Incidencia: 1. Se calcula 1 fq sobre 2000 nacimientos en el mundo. 2. En la raza blanca 1 de cada 20 personas es portadora sana de fq, teniendo una mutación en el cromosoma número 7

50
1. Recién nacidos y lactantes menores Íleo meconial
Criterios de sospecha diagnóstica en pacientes con fibrosis quística de acuerdo a su edad de presentación (OMS, 1995) 1. Recién nacidos y lactantes menores Íleo meconial Ictericia neonatal prolongada (colestásica) Síndrome de edema, anemia, desnutrición Esteatorrea, síndrome de malabsorción Incremento ponderal inadecuado Vómitos recurrentes
 1. Recién nacidos y lactantes menores. Íleo meconial. Ictericia neonatal prolongada (colestásica) Síndrome de edema, anemia, desnutrición. Esteatorrea, síndrome de malabsorción. Incremento ponderal inadecuado. Vómitos recurrentes.")
51
2. Lactantes Tos y/o sibilancias recurrentes o crónicas que no mejora con tratamiento Neumonía recurrente o crónica Retardo del crecimiento Diarrea crónica Prolapso rectal Sabor salado de piel Hiponatremia e hipocloremia crónicas Historia familiar de FQ, o muerte en lactantes o hermanos vivos con síntomas sugerentes

52
3. Preescolares Tos crónica con o sin expectoración purulenta, sin respuesta a tratamiento Sibilancias crónicas recurrentes inexplicadas sin respuesta a tratamiento Incremento deficiente de peso y talla Dolor abdominal recurrente Prolapso rectal Invaginación intestinal Diarrea crónica Hipocratismo digital Hiponatremia e hipocloremia crónicas Hepatomegalia o enfermedad hepática inexplicada Pólipos nasales

53
4. Escolares Síntomas respiratorios crónicos inexplicados Pseudomona aeruginosa en secreción bronquial Sinusitis crónica, poliposis nasal Bronquiectasias Diarrea crónica Síndrome de obstrucción intestinal distal Pancreatitis Prolapso rectal, hepatomegalia 5. Adolescentes y adultos Enfermedad pulmonar supurativa crónica e inexplicada Hipocratismo digital Dolor abdominal recurrente Cirrosis hepática e hipertensión portal Retardo del crecimiento Esterilidad masculina con azoospermia Disminución de la fertilidad en mujeres

54
Revista chilena de pediatría - Consenso nacional de fibrosis quística
La fibrosis quística (FQ) es una enfermedad hereditaria letal, que se transmite de manera autosómica recesiva. Es más frecuente en los grupos de origen k -
 es una enfermedad hereditaria letal, que se transmite. de manera autosómica recesiva. Es más frecuente en los grupos de origen pid=S &script=sci_arttext - 178k -")
Presentaciones similares
 causada por la bacteria Treponema pallidum. A menudo se le ha llamado “la.>")
>")

















