Descargar la presentación
La descarga está en progreso. Por favor, espere

8
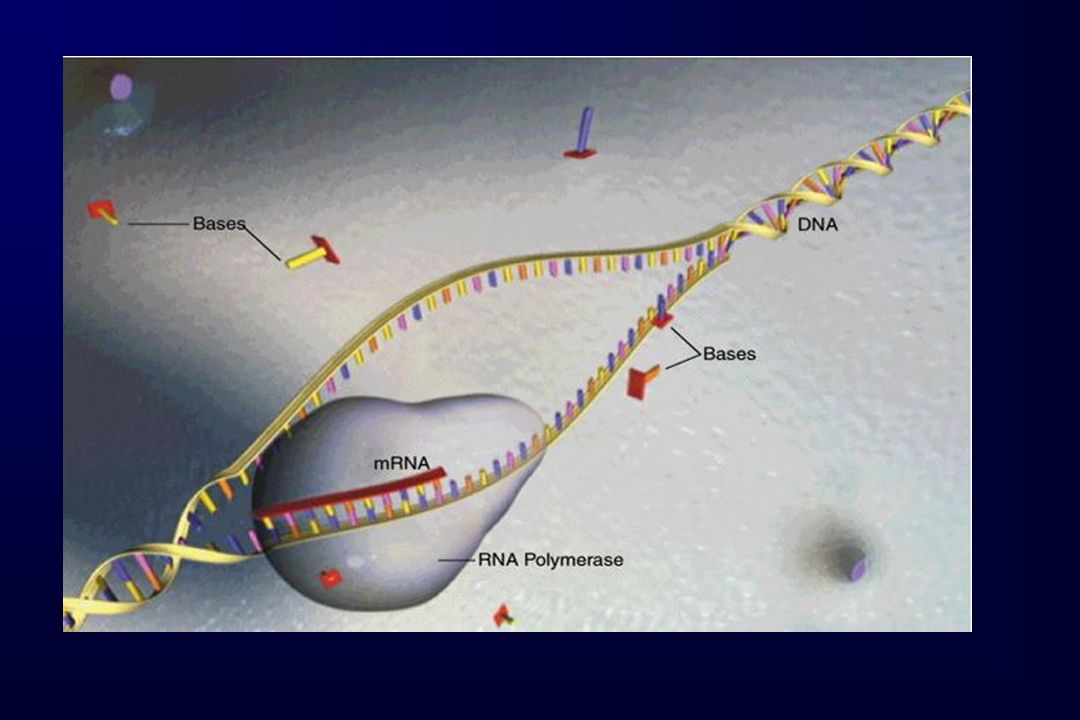
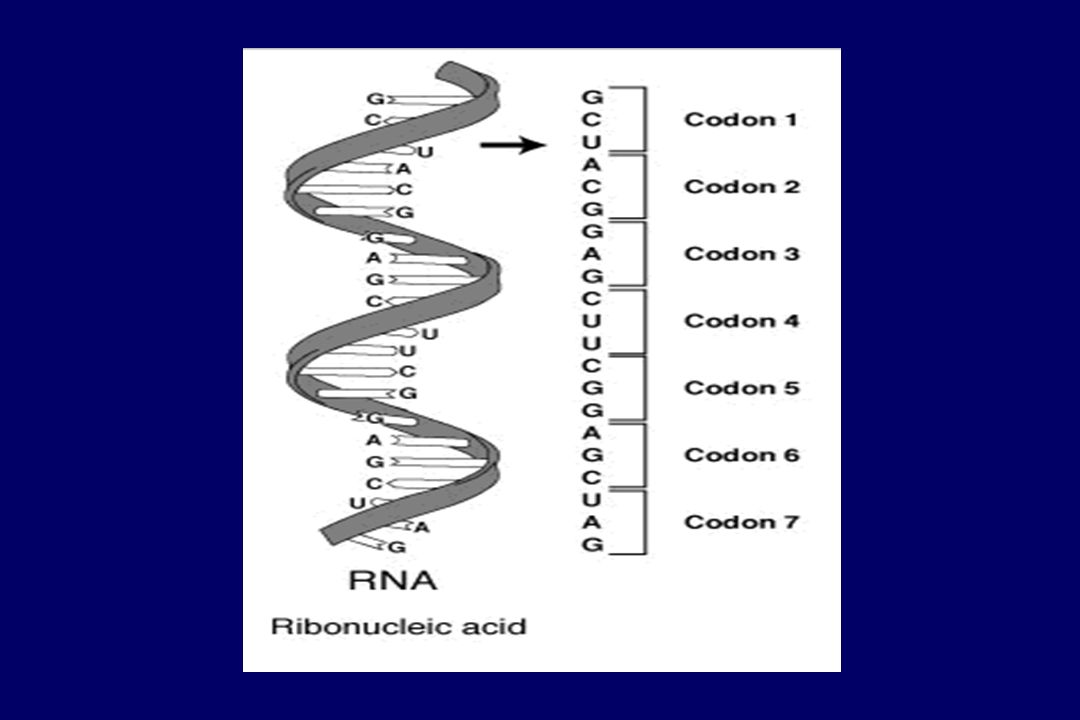
cromosoma DNA RNAm Proteína

10
Variación genética dura vs. blanda o neutra Variación dura > enfermedades genéticas - hereditarias. Gen CFTR: seq. Normal:...ATGACTGTCAGTACGA... Seq. Anormal:...ATGACAGTCAGTACGA... No portador : TT Portador : AT Enfermo: AA AA desarrolla enfermedad independiente. del ambiente Variación blanda > susceptibilidad a sufrir una enfermedad según el ambiente. Gen APOE: seq. E3.. ACGTACGTACAGTACGATACG... Seq. E4.. ACGTACCTACAGTACGATACG... En una dieta alta en grasas (eg Finlandia), los E4E4 y E3E4 tienen mayor riesgo de sufrir un infarto que los E3E3 En Asturias esta variación no está asociada al infarto
, los E4E4 y E3E4 tienen mayor riesgo de sufrir un infarto que los E3E3 En Asturias esta variación no está asociada al infarto.")
11
DIAGNÓSTICOS – Enfermedades hereditarias/monogénica Identificación de mutación responsable de la patología Diagnóstico genético confirmativo Diagnóstico presintomático Diagnóstico prenatal PRONÓSTICOS (susceptibilidad/cribado)- Factores de riesgo asociado a susceptibilidad uso clínico limitado investigación Farmacogenéticos TEST GENÉTICOS
- Factores de riesgo asociado a susceptibilidad uso clínico limitado investigación Farmacogenéticos TEST GENÉTICOS")
12
enfermedades con base genética detección de la mutación responsable de la patología MUTACIONES

13
Madre Enferma Padre sano Hijo enfermo Hijo sano 50% 50% HERENCIA AUTOSÓMICA DOMINANTE

14
Madre Portadora asintomática Padre portador asintomático Hijo/a sano Portadora/portador asintomático Hijo/a enfermo 25% 50% 25% HERENCIA AUTOSÓMICA RECESIVA

15
Madre Portadora obligada Padre sano Hijo sano Hija sana Hija portadora Hijo enfermo 25% 25% 25% 25% HERENCIA LIGADA AL CROMOSOMA X

16
GENÉTICA ESTUDIOS FUNCIONALES NEUROFISIOLOGÍA ANATOMIA PATOLÓGICA NEUROIMAGEN DIAGNÓSTICOS MÁS PRECISOS Y FIABLES ENFERMEDAD NEUROLÓGICA

17
NEUROGENÉTICA : TESTS DIAGNÓSTICOS GEN X/ LOCUS ANÁLISIS MOLECULAR DIRECTO (FIABILIDAD 100%)/INDIRECTO (PROBABILIDAD) IDENTIFICACIÓN DE LA MUTACIÓN /LIGAMIENTO POSITIVO DESCARTAR O CONFIRMAR DIAGNÓSTICO
/INDIRECTO (PROBABILIDAD) IDENTIFICACIÓN DE LA MUTACIÓN /LIGAMIENTO POSITIVO DESCARTAR O CONFIRMAR DIAGNÓSTICO")
18
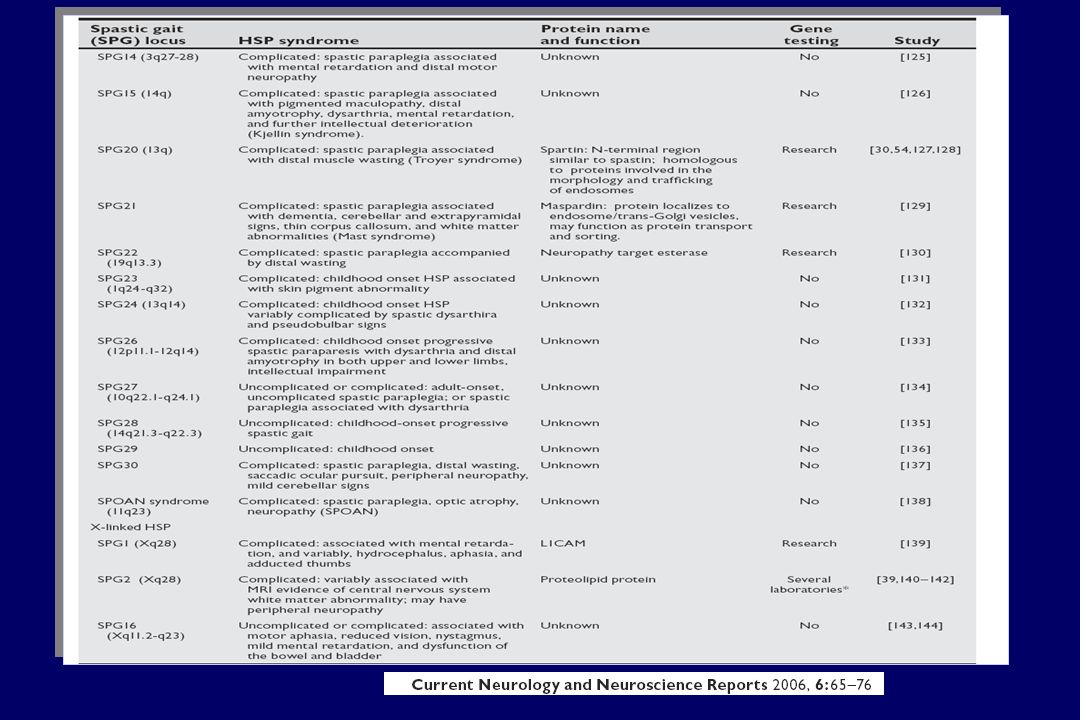
Degeneración retrógrada de los axones largos del tracto corticoespinal Paresia y espasticidad progresivas en los miembros inferiores Prevalencia: 1.27-9.6 de 100.000 PEH pura Síntomas piramidales Debilidad y espasticidad bilateral en MMI Hiperreflexia de MMII Respuesta de extensión plantar Urgencia urinaria; deterioro de sentido vibratorio PARAPARESIS ESPÁSTICA HEREDITARIA (PEH) PEH complicada Síntomas adicionales Retraso mental;Ataxia;Amiotrofia;Sordera ;Epilepsia
 PEH complicada Síntomas adicionales Retraso mental;Ataxia;Amiotrofia;Sordera ;Epilepsia")
19
PARAPARESIS ESPÁSTICA HEREDITARIA (PEH) l Grupo de enfermedades heterogéneas clínica y genéticamente l Gran variabilidad en curso clínico, edad de comienzo, progresión mejor pronóstico edades tempranas (< 30 años) l variabilidad entre los diferentes tipos genéticos de PSH l variabilidad entre diferentes familias con mismo tipo genético correlación genotipo-fenotipo l variabilidad entre miembros de la misma familia familias con mutación en SPG4 en las que coexisten individuos con inicio en la infancia e individuos con comienzo > 30 años factores moduladores (genéticos y/o ambientales) mosaicismo somático (SPG4)
 l Grupo de enfermedades heterogéneas clínica y genéticamente l Gran variabilidad en curso clínico, edad de comienzo, progresión mejor pronóstico edades tempranas (< 30 años) l variabilidad entre los diferentes tipos genéticos de PSH l variabilidad entre diferentes familias con mismo tipo genético correlación genotipo-fenotipo l variabilidad entre miembros de la misma familia familias con mutación en SPG4 en las que coexisten individuos con inicio en la infancia e individuos con comienzo > 30 años factores moduladores (genéticos y/o ambientales) mosaicismo somático (SPG4)")
20
GENÉTICA PEH Más de 30 loci asociados Tipo de Herencia A. Dominante ( ~ 80% ) A. Recesiva ( ~15% ) ( ~ 15% ) Ligada al X ( ~5% ) ( ~ 5% ) SPG3A atlastina (~ 10%) SPG4 espastina (~ 40%) SPG6 NIPA1 SPG 31 REEP1 SPG7 paraplegina Características clínicas similares en muchas de las formas (SPG3A, SPG4, SPG6 o SPG8) Pocas formas con síntomas clínicos específicos (Síndrome de Troyer SPG20 o Síndrome de Silver SPG17
 A. Recesiva ( ~15% ) ( ~ 15% ) Ligada al X ( ~5% ) ( ~ 5% ) SPG3A atlastina (~ 10%) SPG4 espastina (~ 40%) SPG6 NIPA1 SPG 31 REEP1 SPG7 paraplegina Características clínicas similares en muchas de las formas (SPG3A, SPG4, SPG6 o SPG8) Pocas formas con síntomas clínicos específicos (Síndrome de Troyer SPG20 o Síndrome de Silver SPG17.")
21
K

23
No existe un único mecanismo fisiopatogénico asociado al desarrollo de PEH genes mutados codifican proteínas con funciones muy diferentes anomalías en el transporte axonal y el citoesqueleto (SPG10 ( KIF5A); SPG4 (espastina) transporte de vesiculas a traves de los sistemas membranosos intracelulares (SPG3A, Atlastina) disfunción del sistema mitocondrial (SPG7, paraplejina) anomalías en el desarrollo del tracto corticoespinal (SPG1, LICAM)
; SPG4 (espastina) transporte de vesiculas a traves de los sistemas membranosos intracelulares (SPG3A, Atlastina) disfunción del sistema mitocondrial (SPG7, paraplejina) anomalías en el desarrollo del tracto corticoespinal (SPG1, LICAM)")
24
PSH tipo 4 - SPG4 l gen SPG4 l cromosoma 2p;17exones; autosómico dominante l más de 200 mutaciones l es la forma más frecuente de PSH l 40% de las formas dominantes l 10-12% formas esporádicas edad media de inicio entre 26-35 años Rango de edad 2-70 años No correlación entre tipo de mutación y edad de comienzo l forma usualmente no complicada o algunas veces complicada con DCO u otras alteraciones neurológicas l Familias con deterioro cognitivo l Familias con PS, retraso mental, trastornos auditivos, defectos en cuerpo calloso, y atrofia cerebelosa l variabilidad clínica condicionada por polimorfismos en el gen SPG4 (factores moduladores ) l
 l")
25
AAA proteínas proteínas con dominios adenosin trifosfato: unen e hidrolizan ATP Paraplejina (SPG7)( mitocondrial) ; katatina diversas actividades celulares ciclo celular; transporte de vesiculas (axones);proteolísis función de la mitocondria Espastina salvaje interacciona con el microtubulo induce despolimerización ; unión transitoria a través de extremo amino regulada por dominio AAA Espastina mutante se une de modo permanente reorganización del esqueleto patrón filamentoso ovillos perinucleares gruesos pérdida del aster Familia de las AAA proteínas
( mitocondrial) ; katatina diversas actividades celulares ciclo celular; transporte de vesiculas (axones);proteolísis función de la mitocondria Espastina salvaje interacciona con el microtubulo induce despolimerización ; unión transitoria a través de extremo amino regulada por dominio AAA Espastina mutante se une de modo permanente reorganización del esqueleto patrón filamentoso ovillos perinucleares gruesos pérdida del aster Familia de las AAA proteínas")
26
Dominio AAA 342 599 616 Mutaciones missense 789 10 11 12 13 14 15 16 SPG4 Espastina mutaciones en el dominio AAA interfieren con la unión e hidrólisis del ATP

27
Espastina?? Espastina Severing protein?

28
SPG3A/ATLASTINA 10% de las formas dominantes formas puras con edades de inicio tempranas (edad media de inicio <10 años ), y progresión lenta gen con 14 exones y se expresa fundamentalmente en SNC 30 mutaciones en dominios funcionales pérdida o ganancia de función proteína atlastina (558 aminoácidos) pertenece a la familia de las guanidin trifosfatasas (GTPases) 2 dominios transmembrana/GTPasas implicadas en tráfico intracelular Expresión ubicua pero mayor en regiones del cerebro que controlan el movimiento voluntario Atlastina mutada RE-AG afectado desarrollo o función axonal inadecuados
, y progresión lenta gen con 14 exones y se expresa fundamentalmente en SNC 30 mutaciones en dominios funcionales pérdida o ganancia de función proteína atlastina (558 aminoácidos) pertenece a la familia de las guanidin trifosfatasas (GTPases) 2 dominios transmembrana/GTPasas implicadas en tráfico intracelular Expresión ubicua pero mayor en regiones del cerebro que controlan el movimiento voluntario Atlastina mutada RE-AG afectado desarrollo o función axonal inadecuados")
29
Arch Neurol 2004;61:1867-72

30
129 pacientes españoles con PEH esporádica o AD –Procedentes de diferentes puntos de España Extracción de ADN /Secuenciación automática de los genes SPG4, SPG3A (NIPA1, REEP1) »Gen SPG4 todos los pacientes »Gen SPG3A pacientes AD y/o edad de inicio inferior a 10 años »Pacientes con formas AD negativos SPG4 /SPG3A->REEP1, NIPA1 »En proceso, estudio de formas recesivas SPG7/paraplejina Pocas mutaciones recurrentes/Mutaciones privadas Nuevas mutaciones: »estudio de controles sanos »Segregación familiar »conservación de aminoácido ESTUDIO MOLECULAR DE LOS GENES ASOCIADOS A PEH
 »Gen SPG4 todos los pacientes »Gen SPG3A pacientes AD y/o edad de inicio inferior a 10 años »Pacientes con formas AD negativos SPG4 /SPG3A->REEP1, NIPA1 »En proceso, estudio de formas recesivas SPG7/paraplejina Pocas mutaciones recurrentes/Mutaciones privadas Nuevas mutaciones: »estudio de controles sanos »Segregación familiar »conservación de aminoácido ESTUDIO MOLECULAR DE LOS GENES ASOCIADOS A PEH")
31
Casos índice 26/117 casos índice portadores (22%) »42% (22/52 ) de pacientes AD portadores »6%(4/64) de pacientes esporádicos Distribución de mutaciones Distribución de mutaciones »20 / 117 17% en el gen SPG4 »6 / 43 14% en el SPG3A Hombres / Mujeres ( % Hombres )73/56 (55%) Fenotipo puro / complicado123 (95%) / 6 (5%) Duraci ó n (a ñ os) 13 ± 8 Edad de inicio (a ñ os) Media ± D.T. Rango 35 ± 20 1 – 77 Tipo de herencia Esporadico/AF desconocidos:72(56%) Dominante: 56 (43%) Recesivo: 1((1%)
 Dominante: 56 (43%) Recesivo: 1((1%).")
32
Gen SPG4 20 casos índice portadores de mutación en SPG4 –16 AD / 52 31% ( 35-64%) –4 esporádicos / 64 6% ( 10-12%) Pacientes AD incluidos como esporádicos No detectamos deleciones / inserciones de exones Subestimación de la frecuencia de mutaciones en SPG4
 –4 esporádicos / 64 6% ( 10-12%) Pacientes AD incluidos como esporádicos No detectamos deleciones / inserciones de exones Subestimación de la frecuencia de mutaciones en SPG4")
33
A Mutación Gln347His en el exon 7 del gen SPG4

34
T A T AA G T G CT MUTACION 1340 DEL5 EN EXON 9 DE SPG4

35
MUTACIONES EN SPG4 Glu157LysfsX159 Glu193X C-tN-t MIT AAA-ATPasa Gln347His Leu378Arg Thr463Ala Arg479fs Ala392fsX405Asn405fsAla409Thr delPhe404Glu398fsX406KAVA393-396 5 c.1687+1G/T Ile580Thr A) B) Leu380His C-tN-t MIT AAA-ATPasaAAA-ATPasa 5 c.1687+1G/T Ile580Thr A) B) 1 2 3 4 5 6 7 8 9 10 1112 13 14 15 16 17 17 mutaciones: 41% de mutaciones missense; 41% de mutaciones frameshift Mutaciones descritas previamente I328L P489L Mutaciones nuevas
 B) Leu380His C-tN-t MIT AAA-ATPasaAAA-ATPasa 5 c G/T Ile580Thr A) B) mutaciones: 41% de mutaciones missense; 41% de mutaciones frameshift Mutaciones descritas previamente I328L P489L Mutaciones nuevas")
36
MUTACIONES EN SPG3A 3 Gln154GluArg239CysVal253IleHis256Asn N-t C-t GTPasa TMTM 1 2 3 4 5 67 8 9 10 11 12 1314 Asn440Thr A) B) 3 N-t C-t GTPasa TM TM 14 A) B) 6 casos índice portadores de mutación en el gen SPG3A E.I. media = 4 años (± 4) Todos son AD (6 / 52) 11% del total de AD 5 mutaciones diferentes; todas missense Mutaciones descritas previamente Mutaciones nuevas
 Todos son AD (6 / 52) 11% del total de AD 5 mutaciones diferentes; todas missense Mutaciones descritas previamente Mutaciones nuevas.")
37
SPG-64 Val253Ile ¿ Penetrancia incompleta ? Mutación descrita previamente. Asociada a penetrancia incompleta.

38
FAMILIA SPG19 His256Asn Anticipación génica

39
SPG –57 Gln154Glu C->G EXON 4 MUTACIÓN DE NOVO. MOSAICISMO GERMINAL Elevada frecuencia de mutaciones de novo en SPG3A (25% en algunas series) Pacientes sin AF con edad de inicio <20 años cribado de SPG3A
 Pacientes sin AF con edad de inicio <20 años cribado de SPG3A.")
40
Correlaciones genotipo-fenotipo CARACTERÍSTICAS CLÍNICASSPG3ASPG4p-valor Nº de Familias (portadores)6 (17)20 (36) Asintomáticos (%) 2/15 (26%) 3 no datos 7 (19%)0,492 Fenotipo: puro / complicado100%34/20,641 Edad media al inicio (años)4 ± 441 ± 15<0.001 Edad media al examen / diagnóstico (años)33 ± 2047,3 ± 130,106 Duración media de la enfermedad (años)25 ± 1214 ± 80,038
6 (17)20 (36) Asintomáticos (%) 2/15 (26%) 3 no datos 7 (19%)0,492 Fenotipo: puro / complicado100%34/20,641 Edad media al inicio (años)4 ± 441 ± 15<0.001 Edad media al examen / diagnóstico (años)33 ± 2047,3 ± 130,106 Duración media de la enfermedad (años)25 ± 1214 ± 80,038")
41
Diagnóstico molecular de PEH Valoración clínica Diagnóstico diferencial Arbol genealógico: modo de herencia Abordaje del diagnóstico genético-molecular Asesoramiento genético Diagnóstico presintomático Diagnóstico prenatal

42
CONSEJO GENÉTICO modo de herencia: estimación de riesgo AD riesgo para la descendencia 50% AR riesgo descendencia portadores 100% Ligada al X. Riesgo distinto en función de sexo determinar grado de penetrancia penetrancia dependiente de la edad 85% de los pacientes SPG4 tienen síntomas a los 45 años penetrancia incompleta SPG3A anticipación genética hijos afectados antes que los progenitores (SPG3A) diagnóstico presintomático y prenatal Test genético no predice edad de inicio ni forma de presentación clínica
 diagnóstico presintomático y prenatal Test genético no predice edad de inicio ni forma de presentación clínica.")
43
DIAGNÓSTICO PRESINTOMÁTICO EN ENFERMEDADES NEURODEGENERATIVAS Patologías crónicas, progresivas, de curso irreversible y sin tratamiento Imposible predecir con exactitud como y cuando empezará la enfermedad Planificación de descendencia; eliminar la ansiedad por incertidumbre Protocolo de consejo genético y psicológico

44
DIAGNÓSTICO PRESINTOMÁTICO EN ENFERMEDADES NEURODEGENERATIVAS Equipo multidisciplinar: neurológos, psiquiatras, genetistas Puntos clave prediagnóstico: NO MENORES DE EDAD historia familiar/confirmar diagnóstico informar riesgo descendencia beneficios, limitaciones, y riesgos (efectos adversos) del test. Conocer motivación Privacidad Preparación psicológica del individuo Exploración neurológica Obtención de consentimiento informado

45
DIAGNÓSTICO PRESINTOMÁTICO EN ENFERMEDADES NEURODEGENERATIVAS Elementos Clave Postdiagnóstico: Información confidencial. PRIVACIDAD Acompañante Apoyo psicológico Control de efectos adversos DFT;Ataxias hereditarias; Corea de Huntington 8% de los individuos a riesgo solicitan DP planificación descendencia, económica, eliminar ansiedad pocos efectos adversos

46
DIAGNÓSTICO GENÉTICO PRENATAL Patologías hereditarias. Estudio genético-molecular del progenitor afectado. Mutación caracterizada No indicado en enfermedades poligénicas y mulfifactoriales.

47
Células fetales ADN Amplificación mediante PCR Presencia mutaciónAusencia de mutación Análisis genético-molecular

48
DIAGNOSTICO PREIMPLANTACIONAL Normal/MUTACIÓN SPGNormal/Normal rmal /Mutación No implantación implantación A B

49
FAMILIA SPG19 His256Asn CÉLULAS DE VELLOSIDAD CORIÓNICA NO PORTADORAS DE MUTACIÓN EN SPG3A

50
GENETICA MOLECULAR DE LA PEH l conocimiento de la base fisiopatólógica l diagnósticos más precisos l terapias más efectivas farmacogenómica farmacogenética

51
FARMACOGENÓMICA tecnologías genómicas aplicadas al descubrimiento de nuevos farmácos

52
CÓMO SE DESCUBREN LOS FÁRMACOS? Método clásico, empírico, ensayo y error 1. Aislamiento de sustancias de plantas, animales, etc. 2. Probar el efecto de las sustancias en animales de laboratorio: antitumorales?, antirreumáticos?, etc 3. Probar el efecto en pacientes: ensayos clínicos. 4. Describir las bases moleculares por las que la sustancia es eficaz. Método de la Farmacogenómica-Proteómica 1. Descubrir qué genes (=proteínas) contribuyen al origen y/o progresión de una enfermedad. 2. Diseñar y sintetizar fármacos que actúen sobre cada una de esas proteínas.
 contribuyen al origen y/o progresión de una enfermedad. 2. Diseñar y sintetizar fármacos que actúen sobre cada una de esas proteínas..")
53
MUTACION EN GEN X Análisis funcional MAYOR CONOCIMIENTO DELMECANISMO FISIOPATOLÓGICO NUEVAS DIANAS TERAPEUTICAS FARMACOS MÁS EFICACES

54
FARMACOGENÓMICA Y PEH Espastina (SPG4) Hiperestabilizadora de microtubulos Tratamientos con fármacos que desestabilizan microtubulos Nocodazol inhibe el fenotipo mutante en modelos animales de Drosophila SPG4 Futuros estudios en otros modelos animales y ensayos clínicos en humanos.
 Hiperestabilizadora de microtubulos Tratamientos con fármacos que desestabilizan microtubulos Nocodazol inhibe el fenotipo mutante en modelos animales de Drosophila SPG4 Futuros estudios en otros modelos animales y ensayos clínicos en humanos.")
55
NEUROGENÉTICA : TESTS PRONÓSTICOS/FARMACOGENÉTICA GEN X/ LOCUS ANÁLISIS MOLECULAR DE VARIACIÓN TIPO POLIMORFISMO PREDICCIÓN DE RESPUESTA A FARMACO /AJUSTE DE DOSIS PREDICCIÓN DE EFECTOS ADVERSOS 5-10 millones de polimorfismos en el genoma humano respuesta poligénica a fármacos/drogas factores extrínsecos Diferencias poblaciones en frecuencia: importancia de origen poblacional en respuesta farmacológica antihipertensivos : inhibidores de la ECA Polimorfismo ECA I/D intrón 16

56
FARMACOGENETICA.IDENTIFICACIÓN DE DIANAS. Polimorfismos genéticos funcionales Genes de dianas terapeúticas Polimorfismos genéticos de enzimas que participan en la metabolización de fármacos EM de fase I y fase II Respuesta al tratamiento Obviar efectos adversos

57
FARMACOGENÉTICA Y PEH Tratatamiento sintomático; reducción espasticidad Agonista de receptor GABA (baclofeno) Diferente respuesta a baclofeno/ajuste de dosis –Factores genéticos implicados en respuesta (modelos murinos) Identificación de factores genéticos: tratamientos más efectivos/menos efectos adversos
 Diferente respuesta a baclofeno/ajuste de dosis –Factores genéticos implicados en respuesta (modelos murinos) Identificación de factores genéticos: tratamientos más efectivos/menos efectos adversos")
Presentaciones similares









 en el tratamiento del Sindrome Miofascial. Estudio controlado, randomizado, doble ciego con placebo.>")











>")
