Descargar la presentación
La descarga está en progreso. Por favor, espere

1
Mutaciones inestables
Isabel Castro Volio MD, MSc. INISA

2
Sinónimos Mutaciones inestables, mutaciones dinámicas,
Mutaciones por inestabilidad de repeticiones mutaciones por amplificación del ADN, mutaciones por tripletas repetidas, mutaciones por expansiones de trinucleótidos

3
Inestabilidad de repeticiones
Tipo de mutación ligada con más de 40 trastornos Neurológicos Neurodegenerativos Neuromusculares Proceso dinámico de mutación, con productos que continúan mutando dentro de los tejidos y entre las generaciones (inestabilidad somática y germinal)
")
4
El ADN repetitivo Secuencias de ADN no codificante repetidas en tandem: Microsatélites Minisatélites Satélites Megasatélites Otras secuencias repetidas

5
Pequeñas repeticiones de secuencias de ADN
Numerosas en el genoma Localizadas cerca o dentro de unidades de transcripción (genes) Función ? Expansiones de ciertas secuencias repetidas de nucleótidos se asocian a varias patologías hereditarias.
 Función Expansiones de ciertas secuencias repetidas de nucleótidos se asocian a varias patologías hereditarias.")
6
Tipos de microsatélites
También se les llama short tandem repeats (STRs), son repeticiones desde una hasta 6 pares de bases, en tandem
, son repeticiones desde una hasta 6 pares de bases, en tandem.")
7
Tipos de repeticiones asociadas con patología
Trinucleótidos, la unidad repetida son 3 pb: Es la categoría más amplia Tetranucleótidos, 4 pb: Distrofia miotónica tipo 2 (DM2) Pentanucleótidos, 5 pb: Ataxia espinocerebelar 10 (SCA10) Minisatélites, 7-64 pb: Epilepsia progresiva mioclónica 1(EPM1), insulina (INS) Megasatélites varias kb: Distrofia muscular fascioescápulohumeral 1A (FSHND1A)
 Pentanucleótidos, 5 pb: Ataxia espinocerebelar 10 (SCA10) Minisatélites, 7-64 pb: Epilepsia progresiva mioclónica 1(EPM1), insulina (INS) Megasatélites varias kb: Distrofia muscular fascioescápulohumeral 1A (FSHND1A)")
8
Síndrome del cromosoma X frágil

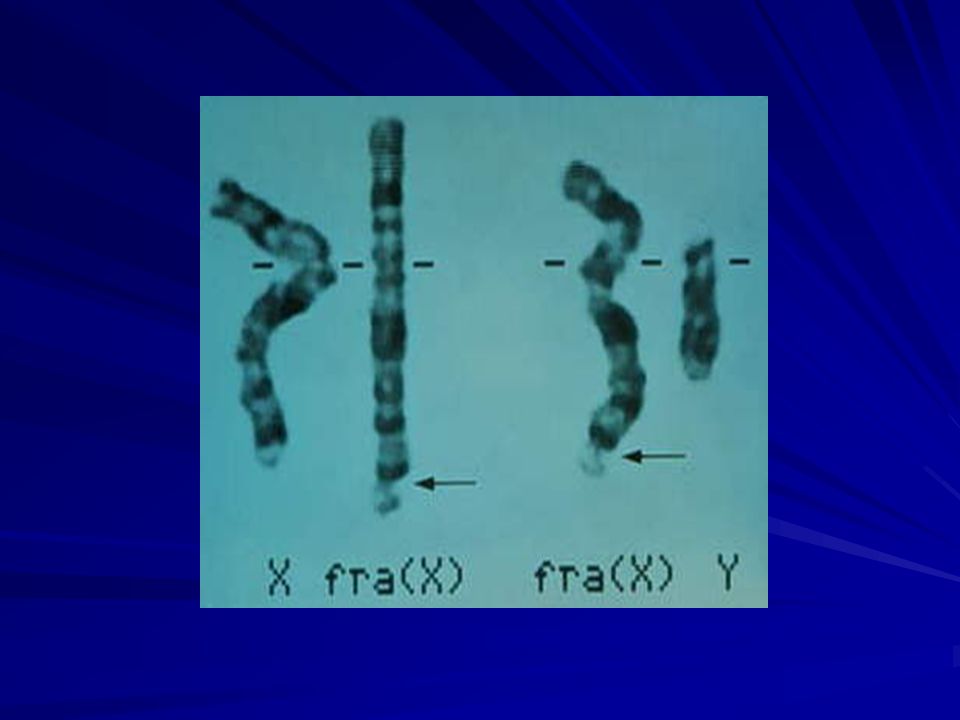
10
El sitio frágil se ubica en Xq27.3

11
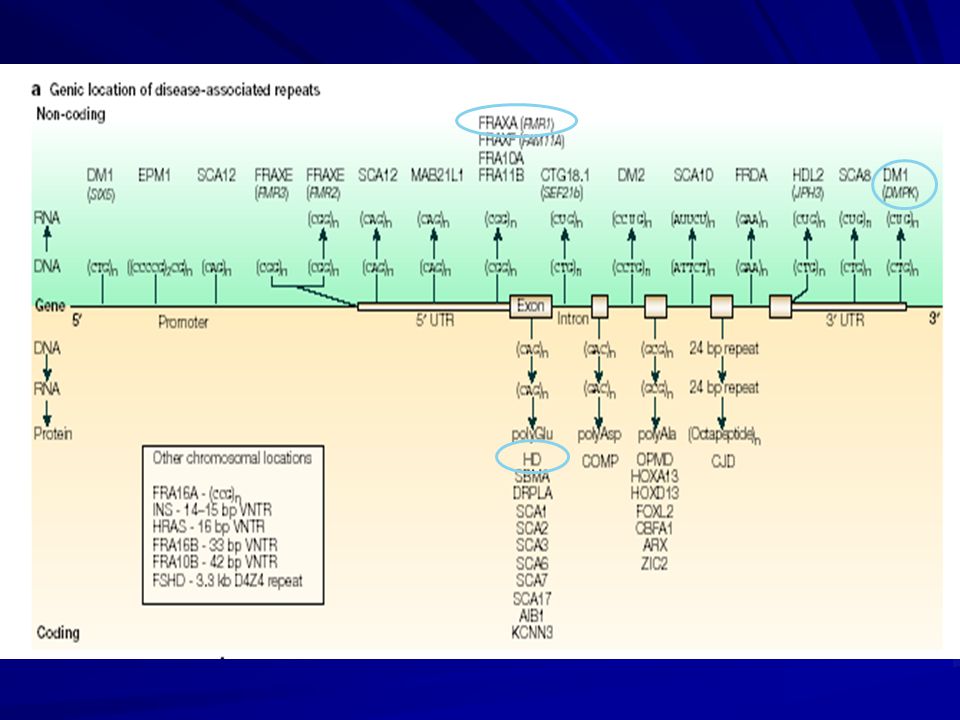
Esquema de la localización de tripletas repetidas en diferentes trastornos genéticos. En rojo las regiones traducidas, en celeste las regiones 5’ y 3’ no traducidas y en azul los intrones. Adaptado de Sinden et al

13
Mutaciones inestables
Síndrome del cromosoma X frágil: primera causa de retardo mental hereditario 6% de la población con retardo mental moderado - severo Distrofia miotónica: principal distrofia muscular del adulto Enfermedad de Huntington, las tres enfermedades son: discapacitantes anticipación genética dominantes requieren terapia génica

16
Síndrome del Cromosoma X Frágil
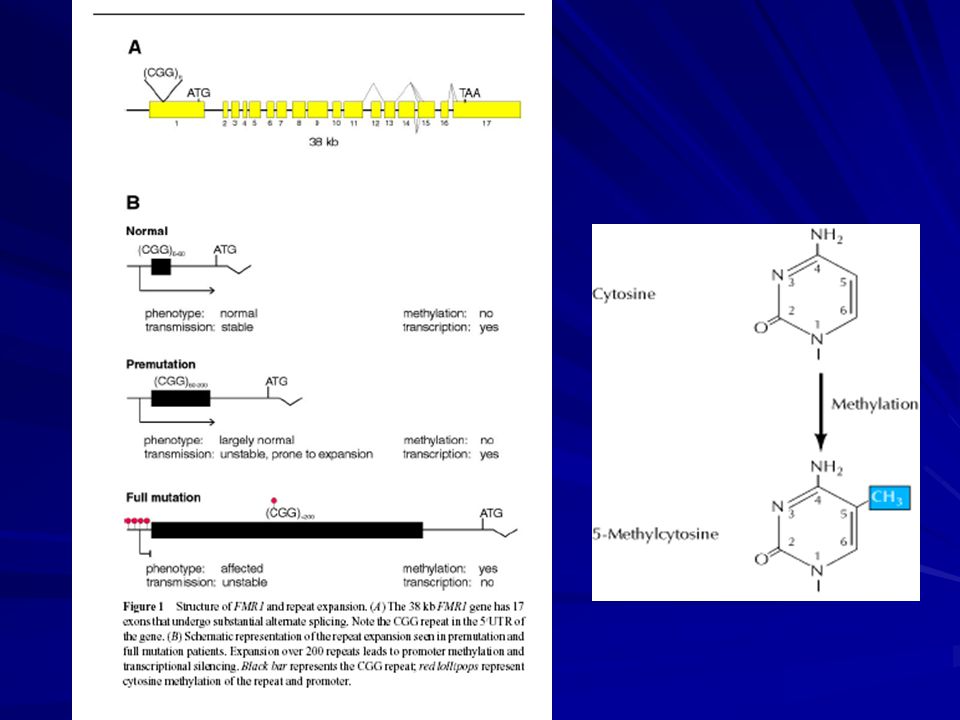
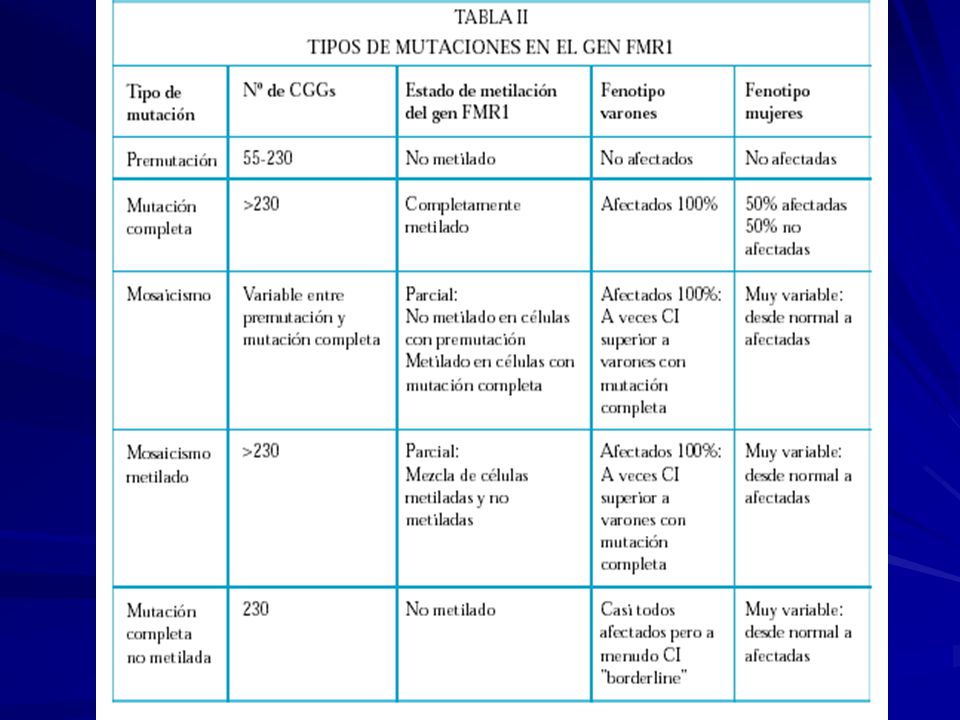
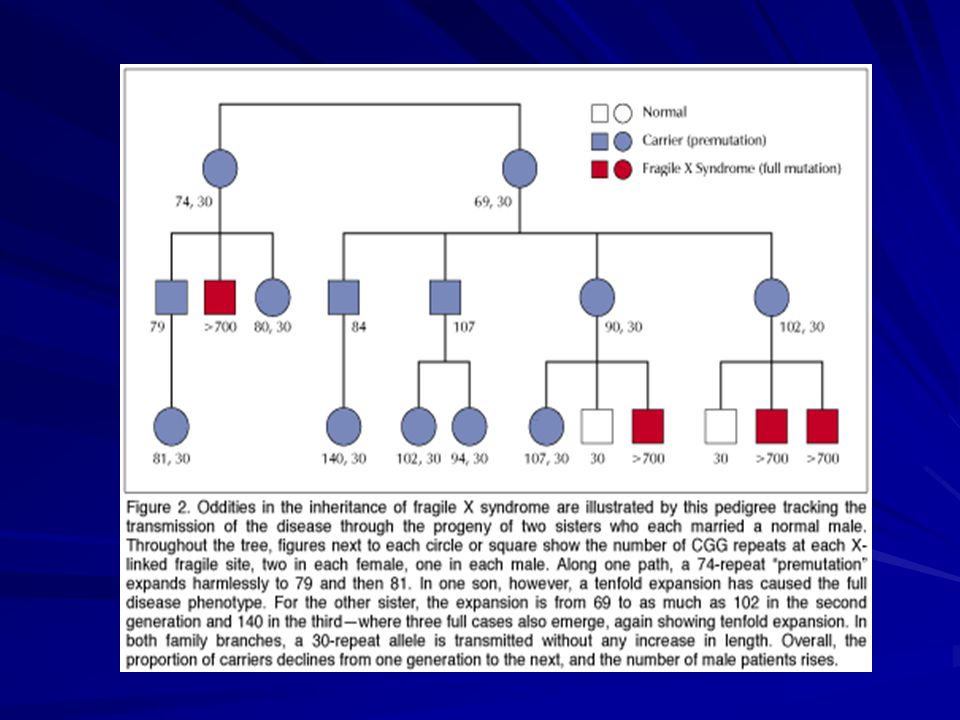
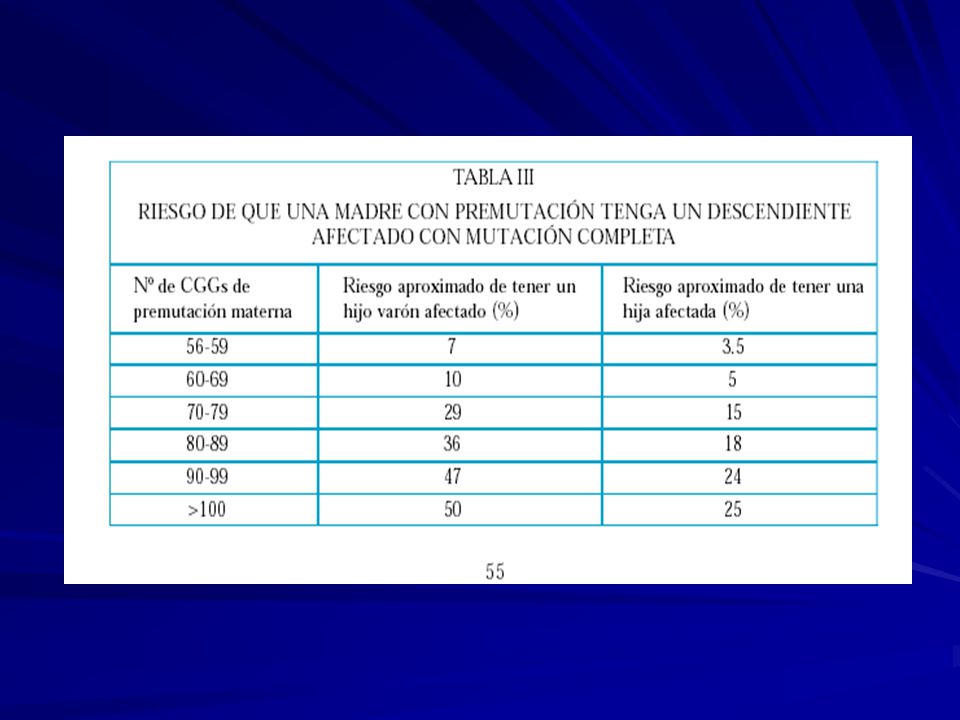
Herencia: dominante ligada al sexo Mutación: en el gen FMR1 Xq27.3 -una expansión de la tripleta CGG en el extremo 5’ no codificante -inestabilidad meiótica y somática. Individuos normales: ~ 5 - ~ 44 repeticiones CGG Alelos intermedios: ~ 45 - ~ 54 Con premutación: ~ 55 - ~ 200 / 230 Pacientes con FRAXA: ~ 201/ 230 o más repeticiones

18
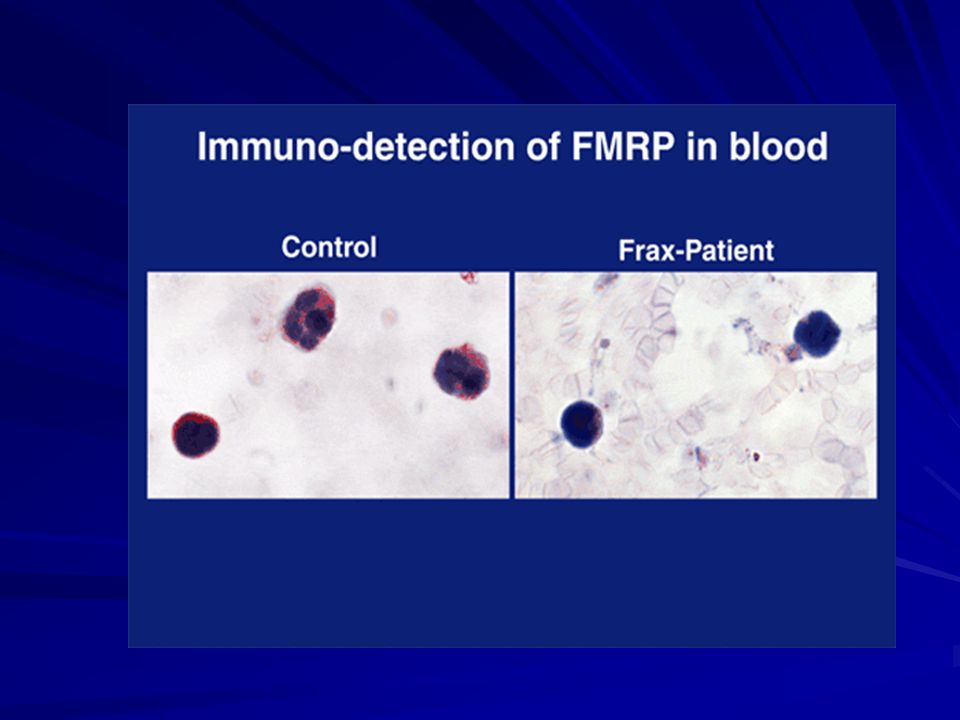
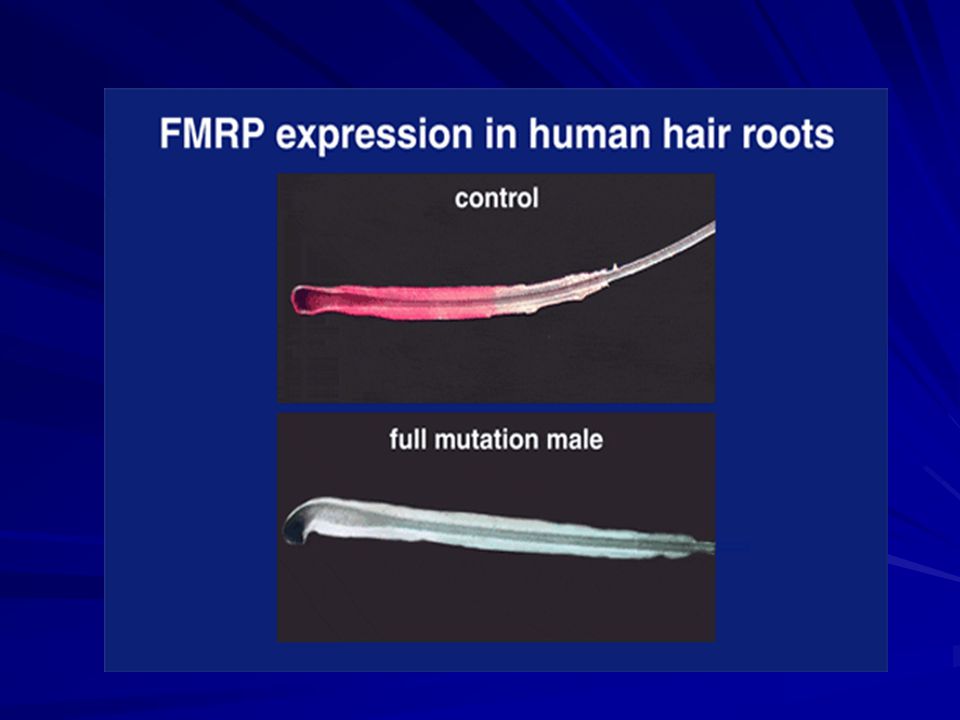
La FMRP = 69 kD

19
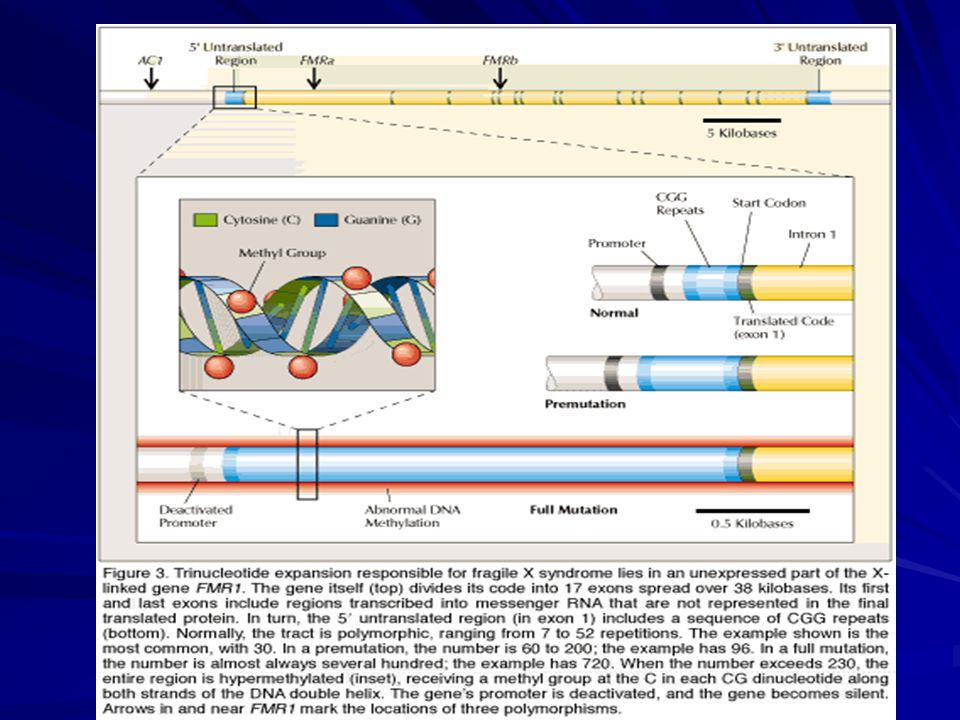
El gen FMR1 abarca 38 kb y consiste de 17 exones.
Codifica un transcripto de kb que se traduce en una proteína de 614 aa. Tiene varios sitios de corte y empalme (splicing) alternativo. El exon 1 es muy rico en CG. La repetición CGG se encuentra dentro del exon 1 en la 5’UTR. 250 bp corriente abajo de la (CGG)n hay una isla CpG que cuando se metila silencia el gen.
 alternativo. El exon 1 es muy rico en CG. La repetición CGG se encuentra dentro del exon 1 en la 5’UTR. 250 bp corriente abajo de la (CGG)n hay una isla CpG que cuando se metila silencia el gen.")
20
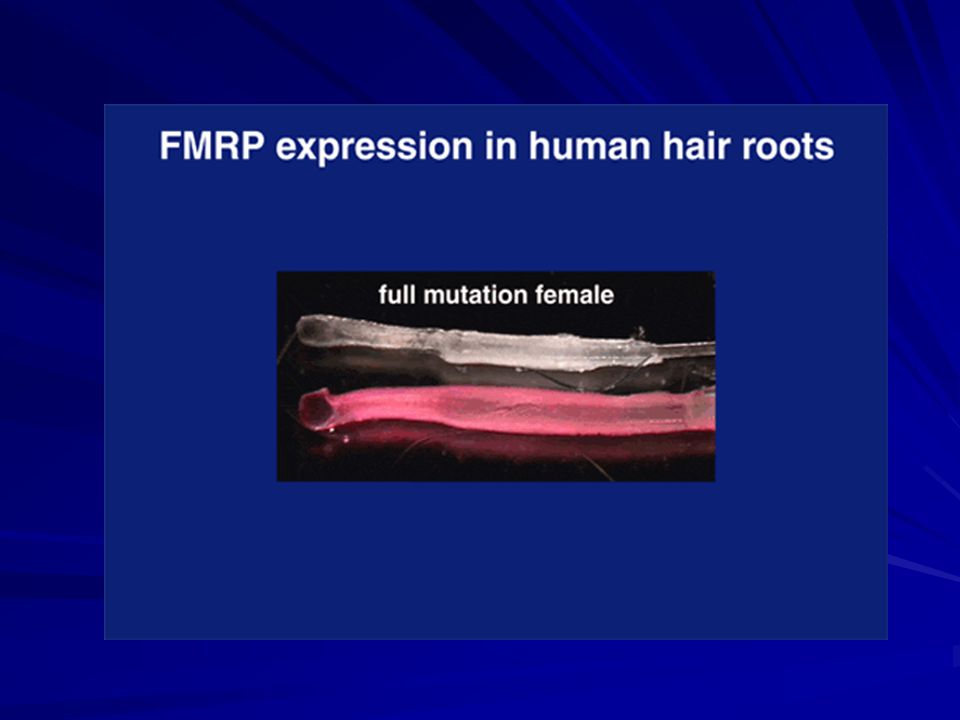
FMRP La proteína FMRP pesa 69 kDa y se producen muchas isoformas debido al splicing alternativo. Se expresa básicamente en el cerebro, específicamente en las neuronas, y en los testículos. FMRP liga ARN selectivamente, forma un complejo de ribonucleoproteína mensajera que se asocia con los poli-ribosomas. Podría estar implicada en la regulación de la traducción a nivel de neuronas via microARNs,

21
FMRP es una proteína que liga ARN y se asocia con los poliribosomas en traducción como parte de una gran ribonucleoproteína mensajera (mRNP) y modula la traducción de sus ligandos al ARN. La FMRP se localiza en las sinapsis y su ausencia altera la plasticidad sináptica, la cual se implica en el aprendizaje y la memoria.

22
Clínica del síndrome del cromosoma X frágil
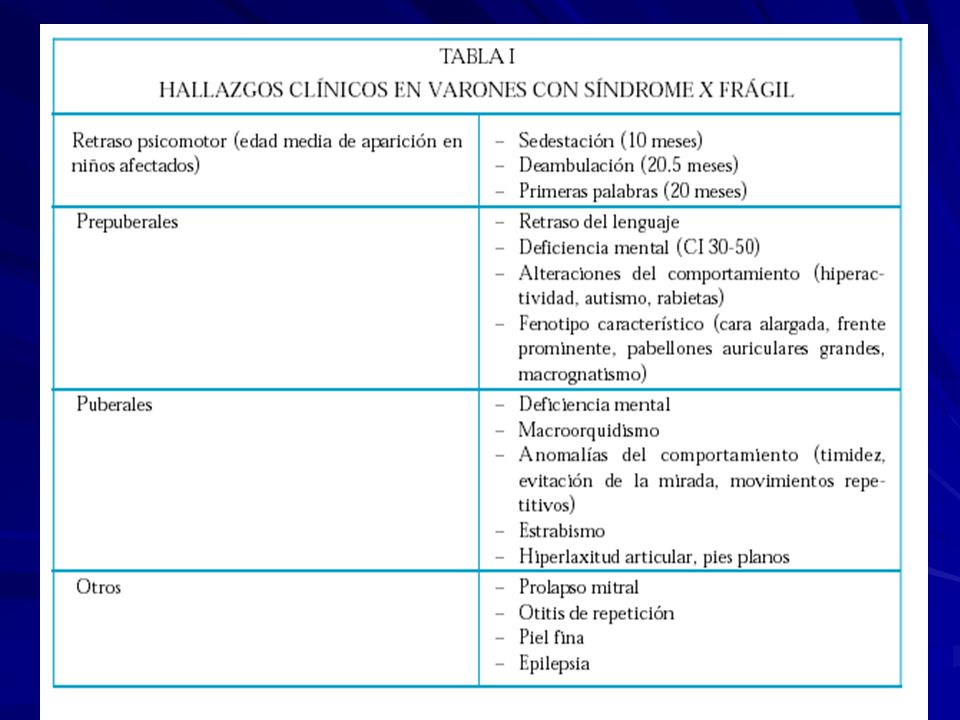
Cara larga y angosta Orejas protuberantes Mentón prominente Macroorquidismo Retardo Mental Hiperactividad Rasgos autistas - aleteo y mordisqueo de las manos - no contacto visual Deficiente integración sensorial Varones 100% afectados, Mujeres < 50% afectadas.

24
FRAXA, cuadro clínico en varones:

26
El fenotipo MARTIN-BELL:
Cara larga y angosta, orejas y mentón prominentes macro-orquidismo

29
Fenotipo en el varón

30
Acentuación del fenotipo

31
PREVALENCIA DEL SÍNDROME X FRÁGIL
Se encuentran casos en todos los grupos étnicos. Afecta 1 : varones afecta 1 : mujeres la prevalencia de la pre-mutación es ~1 : 800 varones ~1 : 250 mujeres

33
Diagnóstico citogenético

34
Diagnóstico del cromosoma X frágil

35
Diagnóstico del cromosoma X frágil
PCR

39
Síndrome del cromosoma X frágil (FRAXA)
1 : varones Genética: - dominante ligada al Cromosoma X - amplificación CGG en el FMR1, Xq27.3 - la mutación completa tiene transmisión materna. - la amplificación causa la inactivación del gen por metilación de la región promotora. - efecto fundador - anticipación genética

42
Lectura obligatoria: emedicine.medscape.com
eMedicine Specialties > Pediatrics: Genetics and Metabolic Disease > Genetics Fragile X Syndrome Jennifer A Jewell, MD, MS, Assistant Professor, Department of Pediatrics, University of Vermont School of Medicine; Pediatric Hospitalist, The Barbara Bush Children's Hospital at Maine Medical Center Updated: Jul 21, 2010

43
Distrofia miotónica Prevalencia 1 : 8 000 en caucásicos
1 : en japoneses 1 : 475 en Quebec, Canadá África, únicamente una familia nigeriana. En Costa Rica, desconocida por el momento.

44
Genética Herencia dominante con expresión variable, penetrancia incompleta y anticipación. Expansión de la tripleta CTG inestable, localizada en el extremo 3’ no traducido del gen DMPK y en la región promotora del gen SIX5 en 19q13.3 Inestabilidad meiótica y somática. Individuos normales: 5-38 repeticiones CTG Pacientes con DM: o más repeticiones

45
Gen DMPK Inicio de Traducción Fin de Traducción Tel Cen 1 2 3 4 5 6 7
8 9 10 11 12 13 14 15 5’ 3’ Intrón 4 Intrón 6 Exón 8 Exón 9 Normal CTG Premutaciones 5-37 rep Afectados 50-80 rep Tel Telómero 100- 2000 rep Cen Centrómero

46
Categorías de pacientes según tamaño de la amplificación en correlación con el fenotipo
I Asintomático II Enfermedad más leve. III Expresión clásica del adulto. IV Expresión pediátrica. V Congénita > repeticiones CTG

47
Cuadro clínico DM1 Atrofia progresiva músculos esqueléticos
Relajación muscular afectada (miotonía) Defectos conducción arritmias cardiacas Cataratas iridiscentes de inicio temprano Resistencia a la insulina e hiperinsulinemia Insuficiencia testicular Calvicie frontal Función alterada del SNC (trastornos cognitivos, somnolencia, trastornos del comportamiento,…)
 Defectos conducción arritmias cardiacas. Cataratas iridiscentes de inicio temprano. Resistencia a la insulina e hiperinsulinemia. Insuficiencia testicular. Calvicie frontal. Función alterada del SNC (trastornos cognitivos, somnolencia, trastornos del comportamiento,…)")
48
Inestabilidad de la mutación
Está directamente relacionada con el tamaño de la amplificación: -repeticiones en el ámbito normal son polimorfismos estables. - si sobrepasan el tamaño umbral de ~ 40 (DM) se vuelven inestables. Es más probable que ocurran expansiones, aunque también se ha descrito la ocurrencia de contracciones. Existe correlación negativa entre edad de expresión y tamaño de la amplificación.
 se vuelven inestables. Es más probable que ocurran expansiones, aunque también se ha descrito la ocurrencia de contracciones. Existe correlación negativa entre edad de expresión y tamaño de la amplificación.")
49
Clínica de la Distrofia Miotónica
Miotonía, desgaste y debilidad muscular progresiva, calvicie, cataratas, problemas respiratorios, arritmias cardiacas, atrofia testicular, resistencia a la insulina. Aparece en el adulto joven, afectando en grado variable los diferentes sistemas, acorta la vida, y es discapacitante. Existe una forma congénita con hipotonía, diplejia facial, retardo mental, defectos en la deglución y succión.

50
Harper,P.S. In: Genetic Inatabilities and Heredetary Neurological Disorders, p.122,1998

51
Distrofia Miotónica Herencia: autosómica dominante con penetrancia
incompleta y expresión variable. Mutación: en el gen DMPK localizado en 19q13.3 -una expansión de la tripleta CTG en el extremo 3’ no codificante -inestabilidad meiótica y somática. Individuos normales: repeticiones CTG Pacientes con DM: o más repeticiones

52
ANTICIPACIÓN

53
Edad y causas de muerte en DM1
Estudio longitudinal en Holanda de 180 pacientes con DM1 clásica : Edad promedio de fallecimiento en varones fue de 59 años, y en mujeres de 60 años. Sobrevivencia observada a los 25 años 99% vs 99% de la pob general 45 años 88% vs 95% 65 años 18% vs 78% Causas de muerte: neumonías y arritmias cardíacas: 73% 50% de los pacientes estaban confinados a la silla de ruedas en el momento de la muerte Otros estudios: esperanza de vida en DMC 35 años.

54
Enfermedad de Huntington (HD)
Descrita por George Huntington en 1872 Sinónimos: Corea de Huntington, mal o baile de San Vito

55
Enfermedad de Huntington (HD)
Enfermedad neurodegenerativa y progresiva del SNC Es uno de los trastornos hereditarios del cerebro más frecuentes Presente en todos los grupos étnicos Prevalencia: - 5-10: en caucásicos - 0.01: en africanos : en japoneses : en chinos -12: Tasmania (Australia) -Región de Zulia (NO Venezuela, junto al lago Maracaibo): 40% de la población está en riesgo, al menos hay 1000 personas mostrando síntomas y otros 3000 más son presintomáticos
 -Región de Zulia (NO Venezuela, junto al lago Maracaibo): 40% de la población está en riesgo, al menos hay 1000 personas mostrando síntomas y otros 3000 más son presintomáticos.")
56
Enfermedad / corea de Huntington
Neurodegenerativa Age of onset promedio a los 40 años años después sobreviene la muerte Afecta ambos sexos por igual. Afecta a todos los grupos étnicos, pero es más frecuente en europeos occidentales (1/10 000). Autosómica dominante. Expansiones de la tripleta CAG
. Autosómica dominante. Expansiones de la tripleta CAG.")
57
Enfermedad de Huntington, clínica
Alteraciones motoras = Corea progresiva Trastorno caracterizado por movimientos rápidos, involuntarios, inútiles, como flexionar y extender los dedos, elevar y descender los hombros o realizar muecas. Progresa hacia movimientos como danzantes que involucran todo el cuerpo. Alteraciones psiquiátricas. Deterioro intelectual, cognitivo.

58
Edad de manifestación HD clásica: 30 y 50 años de edad (manifestación tardía) HD infantil: < 10 años de edad (1-3%) HD juvenil (variante Westphal): < 20 años de edad (5-10%) HD inicio tardío: >50 años de edad (20%) El curso es progresivo, con una evolución media de unos 15-20 años desde el inicio de los síntomas hasta la muerte
: < 20 años de edad (5-10%) HD inicio tardío: >50 años de edad (20%) El curso es progresivo, con una evolución media de unos años desde el inicio de los síntomas hasta la muerte.")
59
Se afectan regiones muy específicas: empieza en el núcleo caudado, continúa expandiéndose hacia el putamen y el globus pallidus. Las neuronas dejan de funcionar y luego mueren, esta pérdida neuronal puede disminuir el peso del cerebro un 25% o hasta un 40%.

60
El gen de la corea de Huntington
Se llama IT15 = Important Transcript 15. Abarca ~ 185 kb de ADN genómico en 4p16.3. Contiene 67 exones, la repetición CAG está en el exon 1. Dos transcriptos principales de ARNm. Codifica la proteína “huntingtina” (htt) La repetición CAG se traduce en una región de poliglutaminas que causa ganancia dominante de función.
 La repetición CAG se traduce en una región de poliglutaminas que causa ganancia dominante de función.")
61
Mutación responsable de la HD
1993: identificación de la alteración genética Expansión del trinucleótido CAG en el exón 1 del gen IT-15 ó HD, cerca del extremo 5’ segmento PoliQ:Huntingtina (htt) Exón 1 Gen IT-15 ó HD Esquema del gen IT-15 ó HD e intervalos según el número de repeticiones CAG Adaptado de Potter et al. 2004
 Exón 1. Gen IT-15 ó HD. Esquema del gen IT-15 ó HD e intervalos según el número de. repeticiones CAG Adaptado de Potter et al")
62
Genética: Mutación Los cromosomas normales poseen de 9 a 35 repeticiones CAG, con una moda de 18 Los cromosomas CH poseen de 36 a 121 repeticiones CAG y la moda es de 48 Se han reconocido 4 intervalos de tamaños de repeticiones CAG asociados con riesgos variables de la enfermedad: A. Normal: 28 repeticiones B. No penetrante con inestabilidad meiótica paterna: 29 a 35 repeticiones C. Penetrancia reducida con inestabilidad meiótica paterna: 36 a 39 repeticiones D. CH: 40 repeticiones Susceptibles a expansión

63
Inestabilidad intergeneracional de la repetición CAG
En la gametogénesis, sobre todo en la transmisión por el varón. En > 80% de las transmisiones de un padre o una madre CH, el # de CAG o en una o pocas unidades. A veces en una transmisión paterna el aumento en el # de repeticiones es muy grande aparición juvenil de la CH (> repeticiones). Mosaicismo en el tamaño de las repeticiones en los espermatozoides de los varones CH.
. Mosaicismo en el tamaño de las repeticiones en los espermatozoides de los varones CH.")
64
Enfermedad de Huntington
Típicamente entre años, a veces entre 2-90 años. Expresión juvenil asociada a transmisión por linaje paterno Casos “esporádicos”: expansiones ocurridas en las meiosis de individuos sanos de sexo masculino con repeticiones. Mutaciones “nuevas” ~ 3%, surgen de alelos paternos intermedios, con repeticiones. Riesgo del 50% Presenta el fenómeno de anticipación genética la huntingtina tiene función ¿?.

65
Ganancia de función tóxica de la htt
No hay CH en deleciones del gen IT15. No hay CH en translocaciones que cortan el gen IT15. El fenotipo de los heteros y los homos es =. Fenotipo dominante. No mutaciones de ninguna clase excepto inestables. Los niveles de htt son = para alelos normales y mutados

66
Neuronas con inclusiones de poliglutamina
Típicas en CH, SCA1, SCA3, SCA7, DRPLA, SBMA. Inclusiones de proteínas con poliglutamina + ubicuitina. Estas inclusiones correlacionan con promoción de la apoptosis. Se llaman inclusiones intranucleares neuronales (NII) pero en CH también hay inclusiones citoplasmáticas.
 pero en CH también hay inclusiones citoplasmáticas.")
67
Lectura obligatoria emedicine.medscape.com
eMedicine Specialties > Neurology > Movement and Neurodegenerative Diseases Huntington Disease Fredy J Revilla, MD, Assistant Professor of Neurology, Head of Division of Movement Disorders, Department of Neurology, University of Cincinnati College of Medicine, Cincinnati Veterans Affairs Medical Center Jaime Grutzendler, MD, Assistant Professor, Department of Neurology and Physiology, Northwestern University School of Medicine; Travis R Larsh, Co-op Student, Department of Neurology, University of Cincinnati College of Medicine Updated: Aug 4, 2010

68
Mutaciones dinámicas, resumen
Sexo de transmisión en la > de enf por repeticiones de CAG, + severa si es paterna, en FRAXA y en DM hay > variación de tamaño en la transmisión materna Inestabilidad somática y germinal Patrones de expresión tisular Mosaicismo y posición dentro de los genes afectados la > de las (CAG)n en región codificante, excepto SCA12 (5’UTR)
n en región codificante, excepto SCA12 (5’UTR)")
Presentaciones similares