Descargar la presentación
La descarga está en progreso. Por favor, espere

1
MPS O GLICOSAMINOGLICANOS (GAG)
MUCOPLISACÁRIDOS MPS O GLICOSAMINOGLICANOS (GAG) -SON MACROMOLÉCULAS FORMADAS POR UNIDADES MONOMÉRICAS DE N-ACETIL-HEXOSAMINA Y ÁCIDO HEXURÓNICO. -Azúcar aminado: glucosamina, galactosamina -Derivado del ácido Urónico ( ac. Hialurónico) SON HETEROPOLISACÁRIDOS CONFORMADOS POR 2 UNIDADES ALTERNANTES Y EN LA QUE POR LO MENOS UNA TIENE UN GRUPO ÁCIDO (COOH) O SULFÚRICO. CUANDO SE ASOCIAN CON PROTEÍNAS MUCINAS O MUCOPROTEÍNAS O PROTEOGLICANOS.
 -SON MACROMOLÉCULAS FORMADAS POR UNIDADES MONOMÉRICAS DE N-ACETIL-HEXOSAMINA Y ÁCIDO HEXURÓNICO. -Azúcar aminado: glucosamina, galactosamina. -Derivado del ácido Urónico ( ac. Hialurónico) SON HETEROPOLISACÁRIDOS CONFORMADOS POR 2 UNIDADES ALTERNANTES Y EN LA QUE POR LO MENOS UNA TIENE UN GRUPO ÁCIDO (COOH) O SULFÚRICO. CUANDO SE ASOCIAN CON PROTEÍNAS MUCINAS O MUCOPROTEÍNAS O PROTEOGLICANOS.")
2
LOS MPS SON: DERMATÁN SULFATO (O CONDROITÍN SULFATO B) KERATÁN SULFATO HEPARÁN SULFATO CONDROITINA A (4-SULFATO) CONDROITINA B (6-SULFATO) FUNCIÓN: LUBRICANTES O COMO CEMENTO INTERCELULAR FLEXIBLE METABOLIZACIÓN: LA DEGRADACIÓN SE PRODUCE DESDE SU EXTREMO NO REDUCTOR EXCRECIÓN: POR ORINA
 FUNCIÓN: LUBRICANTES O COMO CEMENTO INTERCELULAR FLEXIBLE. METABOLIZACIÓN: LA DEGRADACIÓN SE PRODUCE DESDE SU EXTREMO NO REDUCTOR. EXCRECIÓN: POR ORINA.")
3
Células que secretan y/ó producen MPS: Tejido Al acumularse producen:
Sustrato acumulado predominantemente Al acumularse producen: Osteoblastos Hueso Diversos MPS Displasias óseas Fibroblastos Tejido fibroso de córnea, piel, válvulas cardiacas. Sulfato de Keratán Sulfato de Dermatán. Malformaciones de las válvulas cardiacas y opacidad corneal Condroblastos Cartílago Sulfato de Condrointín Displasias de cartílagos Neuronas y linfocitos Tejido nervioso Y sangre Sulfato de heparán Retraso psicomotor

4
ÁCIDO HIALURÓNICO: ES EL MÁS ABUNDANTE PRESENTE EN: SUSTANCIA INTERSTICIAL, TEJIDO EXTRACELULAR CONECTIVO, LÍQUIDO SINOVIAL DE ARTICULACIONES Y HUMOR VÍTREO DEL OJO. QUÍMICAMENTE ES UN POLÍMERO LINEAL, CARGADO (-) A pH 7 ES SOLUBLE EN AGUA. ESTRUCTURA: ÁCIDO HIALURÓNICO HIALURONIDASA_ 2 COMPUESTOS (ÁC. GLUCURÓNICO + GLUCOSAMINA)n (MENOR VISCOCIDAD) CONDROITINA: SIMILAR AL ÁC. HIALURÓNICO: ÁC- D- GLUCURÓNICO + N-AC- D-GALACTOSAMINA ES UN COMPONENTE SECUNDARIO DEL MATERIAL EXTRACELULAR CONDROITINA A 4-SULFATO DE CONDROITINA CONDROITINA B 6-SULFATO DE CONDROITINA COMPONENTES DE CARTÍLAGO, HUESOS, CÓRNEA Y ESTRUCTURAS DEL TEJIDO CONECTIVO DE LOS VERTEBRADOS.
 A pH 7. ES SOLUBLE EN AGUA. ESTRUCTURA: ÁCIDO HIALURÓNICO HIALURONIDASA_ 2 COMPUESTOS. (ÁC. GLUCURÓNICO + GLUCOSAMINA)n (MENOR VISCOCIDAD) CONDROITINA: SIMILAR AL ÁC. HIALURÓNICO: ÁC- D- GLUCURÓNICO + N-AC- D-GALACTOSAMINA. ES UN COMPONENTE SECUNDARIO DEL MATERIAL EXTRACELULAR. CONDROITINA A 4-SULFATO DE CONDROITINA. CONDROITINA B 6-SULFATO DE CONDROITINA. COMPONENTES DE CARTÍLAGO, HUESOS, CÓRNEA Y ESTRUCTURAS DEL TEJIDO CONECTIVO DE LOS VERTEBRADOS.")
5
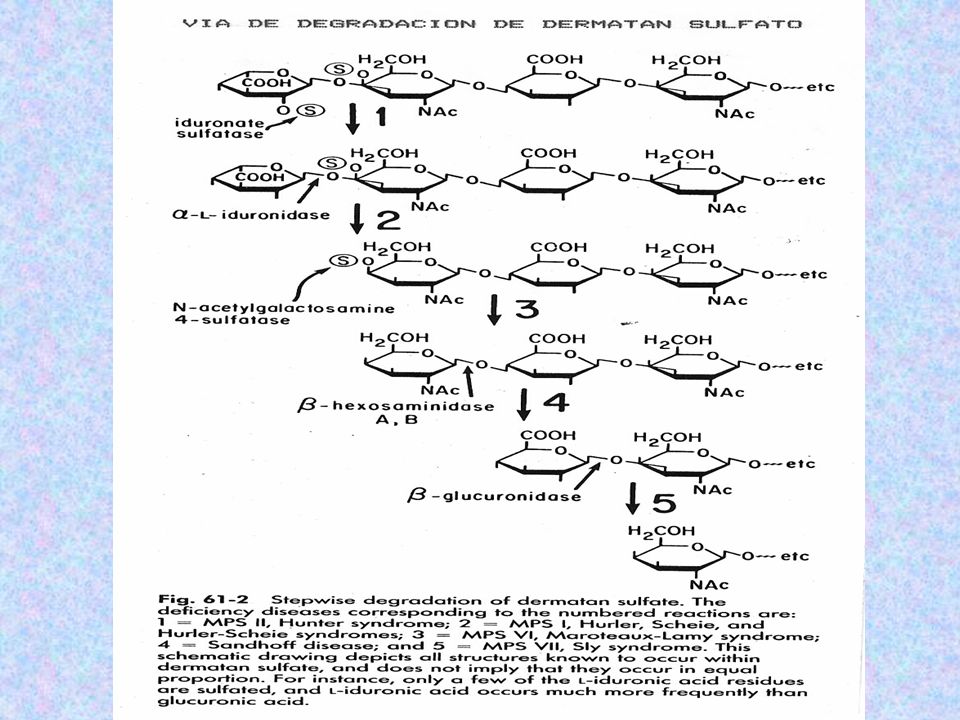
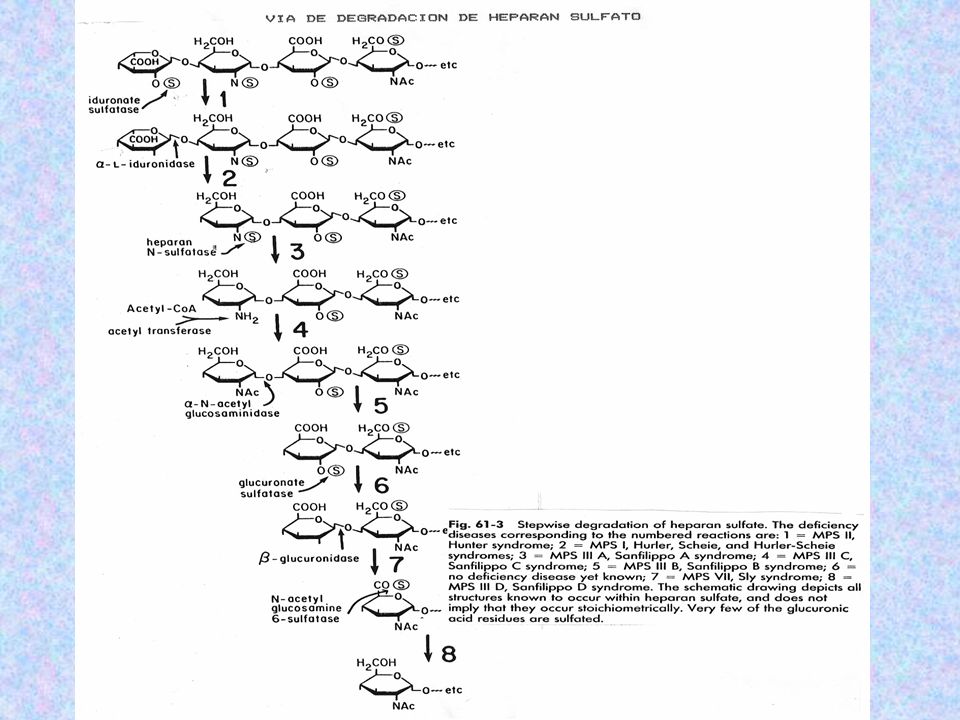
DERMATÁN SULFATO: ÁC. IDURÓNICO Y N-AC-GALACTOSAMINA-4-SULFATO PRESENTE EN PIEL. QUERATÁN SULFATO: GALACTOSA – GALACTOSA-6-SULFATO Y N-AC-GALACTOSAMINA-6- SULFATO COMPONENTE DE TEJIDOS ÓSEOS, CÓRNEAS HEPARINA O HEPARÁN SULFATO: ÁC. IDURÓNICO –ÁC. GLUCURÓNICO-2- SULFATO Y GLUCOSAMINA-6- SULFATO IMPIDE LA COAGULACIÓN SANGUÍNEA, UBICÁNDOSE EN PULMONES Y ARTERIAS.

6
M U C O P O L I S A C A R I D O S I S CLÍNICAMENTE: -SON DESÓRDENES QUE AFECTAN NUMEROSOS ÓRGANOS SE ENCUENTRAN ALTERADAS LAS ENZIMAS LISOSOMALES ENCARGADAS DE LA DEGRADACIÓN DE LOS MPS. ESTO ORIGINA UN INCREMENTO EN LOS NIVELES DE MPS EN LOS LISOSOMAS (DIGERIDOS PARCIALMENTE O NO) LOS MPS MAYORMENTE EXCRETADOS POR ORINA SON: HEPARÁN, DERMATÁN Y QUERATÁN SULFATOS EN DIFERENTES PROPORCIONES SEGÚN LA ENZIMA DEFICIENTE DESÓRDEN: DEFICIENCIA DE HIDROLASAS LISOSOMALES ESPECÍFICAS. CARACTERÍSTICAS CLÍNICAS, GENÉTICAS Y BIOQUÍMICAS: DEPÓSITO PROGRESIVO DE MPS. DEGENERACIÓN ESQUELÉTICA, PROBLEMAS ARTICULARES, LIMITACIÓN EN EL MOVIMIENTO ENTURBIAMIENTO DE LAS CÓRNEAS, ALTERACIONES CARDÍACAS HEPATOESPLENOMEGALIA FACIE GROTESCA CABEZA GRANDE, ABDOMEN PROMINENTE
 LOS MPS MAYORMENTE EXCRETADOS POR ORINA SON: HEPARÁN, DERMATÁN Y QUERATÁN SULFATOS EN DIFERENTES PROPORCIONES SEGÚN LA ENZIMA DEFICIENTE. DESÓRDEN: DEFICIENCIA DE HIDROLASAS LISOSOMALES ESPECÍFICAS. CARACTERÍSTICAS CLÍNICAS, GENÉTICAS Y BIOQUÍMICAS: DEPÓSITO PROGRESIVO DE MPS. DEGENERACIÓN ESQUELÉTICA, PROBLEMAS ARTICULARES, LIMITACIÓN EN EL MOVIMIENTO. ENTURBIAMIENTO DE LAS CÓRNEAS, ALTERACIONES CARDÍACAS. HEPATOESPLENOMEGALIA. FACIE GROTESCA. CABEZA GRANDE, ABDOMEN PROMINENTE.")
7
CAUSA DE MUERTE: -INFECCIONES RESPIRATORIAS, TRASTORNOS CARDÍACOS OTRAS CARACTERÍSTICAS CLÍNICAS SORDERA MANOS EN GARRA COLUMNA VERTEBRAL REDONDEADA BRAZOS LARGOS Y PIERNAS CORTAS OJOS CON ÓRBITAS GRANDES, HIDROCEFALIA GENÉTICA: AUTOSÓMICA RECESIVA INCIDENCIA: 1/ – PORTADORES 1/ 150 DEPÓSITO DE MPS: SEMEJAN MATERIAL DE INCLUSIÓN, AL MICROSCOPIO ELECTRÓNICO SE OBSERVAN CON ASPECTO FINAMENTE GRANULAR EN LISOSOMAS EN NEURONAS HAY INCLUSIONES LISOSOMALES DEPÓSITO DE MPS SOBRE EL REVESTIMIENTO DE ARTERIAS CORONARIAS PSEUDOATEROMATOSIS AL AUMENTAR LOS MPS HAY MAYOR PRODUCCIÓN DE COLÁGENO ENTUMECIMIENTO ARTICULAR.

8
DIAGNÓSTICO: MUCOPOLISACARIDURIA: MUY MARCADA DETECCIÓN EN ORINA DE MPS MÉTODOS: AZUL DE TOLUIDINA (METACROMACIA) ALBÚMINA ÁCIDA (TURBIDEZ) SALES DE CETILPIRIDINIUM (TURBIDEZ) RELACIÓN DERMATÁN/ HEPARÁN = 7: 3 MEDICIÓN DE LA AE (CULTIVO DE FIBROBLASTOS) USANDO COMO SUSTRATO FENIL-IDURONATO DE SODIO (AE EN HETEROCIGOTAS ES ½)
 SALES DE CETILPIRIDINIUM (TURBIDEZ) RELACIÓN DERMATÁN/ HEPARÁN = 7: 3. MEDICIÓN DE LA AE (CULTIVO DE FIBROBLASTOS) USANDO COMO SUSTRATO FENIL-IDURONATO DE SODIO. (AE EN HETEROCIGOTAS ES ½)")
9
MUCOLIPIDOSIS: DESÓRDEN CON CARACTERÍSTICAS CLÍNICAS Y BIOQUÍMICAS SIMILARES A LAS MPS Y ESFINGOLIPIDOSIS CLASIFICACIÓN: MUCOLIPIDOSIS II O ENFERMEDAD DE LAS CÉLULAS I MUCOLIPIDOSIS III O PSEUDOHURLER O POLIDISTROFIA DEFICIENCIAS MÚLTIPLE DE SULFATASAS

11
MPS TIPO I O SÍNDROME DE HURLER:
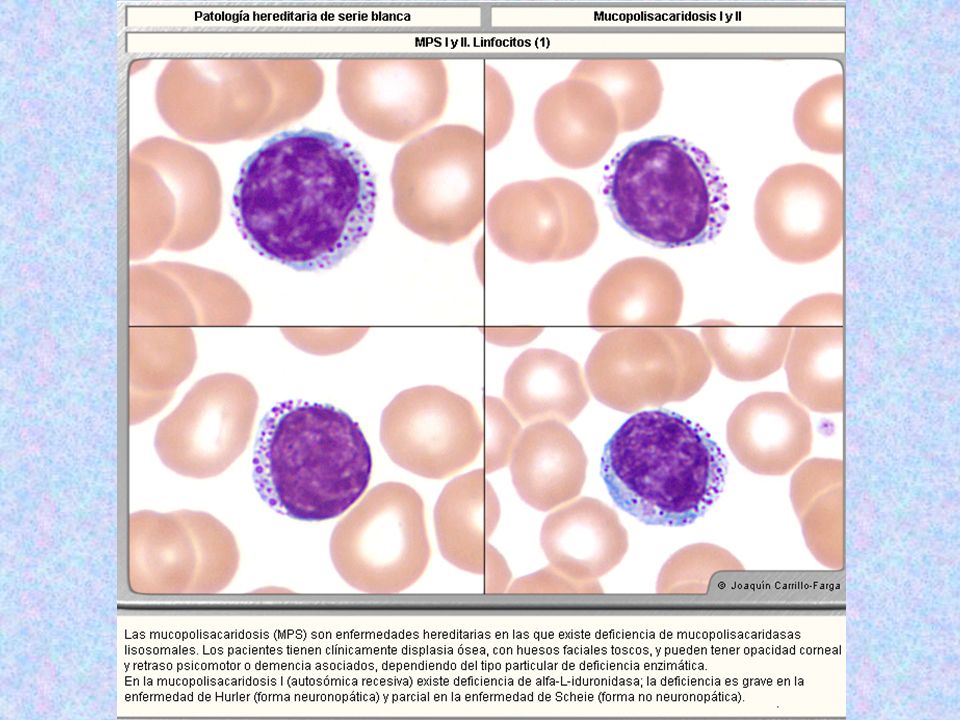
MPS I H DEFECTO: ALFA- L- IDURONIDASA (SÍNDROME DE HURLER Y SCHEIE) SE ACUMULAN RESIDUOS DE ÁC.URÓNICO (DERMATÁN Y HEPARÁN) TÍPICA ALTERACIÓN CON DEPÓSITO DE MPS DESÓRDENES PROGRESIVOS Y SEVEROS (MUERTE A LOS 10 AÑOS) CARACTERÍSTICAS CLÍNICAS ENTURBIAMIENTO CORNEAL, LORDOSIS LUMBAR, ENTUMECIMIENTO ARTICULAR INFANCIA DETERIORO PROGRESIVO (MENTAL Y FÍSICO CON ENANISMO A 2-3 AÑOS) DETENCIÓN DEL CRECIMIENTO DIAGNÓSTICO: ACUMULACIÓN 35S – MPS EN CULTIVOS DE FIBROBLASTOS DIAGNÓSTICO PRENATAL A PARTIR DEL SEGUNDO TRIMESTRE DE VIDA FETAL LAS CÉLULAS ANMIÓTICAS TIENEN SIMILAR COMPORTAMIENTO QUE LOS FIBROBLASTOS DE ADULTOS
 SE ACUMULAN RESIDUOS DE ÁC.URÓNICO (DERMATÁN Y HEPARÁN) TÍPICA ALTERACIÓN CON DEPÓSITO DE MPS. DESÓRDENES PROGRESIVOS Y SEVEROS (MUERTE A LOS 10 AÑOS) CARACTERÍSTICAS CLÍNICAS ENTURBIAMIENTO CORNEAL, LORDOSIS LUMBAR, ENTUMECIMIENTO ARTICULAR. INFANCIA DETERIORO PROGRESIVO (MENTAL Y FÍSICO CON ENANISMO A 2-3 AÑOS) DETENCIÓN DEL CRECIMIENTO. DIAGNÓSTICO: ACUMULACIÓN 35S – MPS EN CULTIVOS DE FIBROBLASTOS. DIAGNÓSTICO PRENATAL A PARTIR DEL SEGUNDO TRIMESTRE DE VIDA FETAL. LAS CÉLULAS ANMIÓTICAS TIENEN SIMILAR COMPORTAMIENTO QUE LOS FIBROBLASTOS DE ADULTOS.")
12
MPS I S O SÍNDROME DE SCHEIE
DEFECTO: DEFICIENCIA DE ALFA-L- IDURONIDASA, TIPO SCHEIE GENÉTICA: AR DOS VARIANTES ENZIMÁTICAS (CROMATOGRAFÍA DEAE-CELULOSA) SÍNDROME S (NO ADSORCIÓN SOBRE DEAE TERMOLÁBIL) SÍNDROME H (ADSORCIÓN SOBRE DEAE TERMOLÁBIL A LOS 56° C) AMBAS POSEEN pH ÓPTIMO Y Km SIMILARES MANIFESTACIONES CLÍNICAS: SEVERO ENTURBIAMIENTO CORNEAL + DEGENERACIÓN PIGMENTACIÓN DE LA RETINA INCAPACIDAD VISUAL DEFORMACIÓN EN MANOS, FACIE GROTESCA, BOCA ABIERTA ENTUMECIMIENTO ARTICULAR POSIBLE DISTURBIO SIQUIÁTRICO ALTERACIONES VÁLVULA AÓRTICA (ESTENOSIS O REGURGITACIÓN) INTELIGENCIA NORMAL SNC NEURONAS CORTICALES NORMALES MPS LIGERAMENTE AUMENTADOS RESPECTO A MPS IH LESIONES EN HÍGADO Y BAZO
 SÍNDROME S (NO ADSORCIÓN SOBRE DEAE TERMOLÁBIL) SÍNDROME H (ADSORCIÓN SOBRE DEAE TERMOLÁBIL A LOS 56° C) AMBAS POSEEN pH ÓPTIMO Y Km SIMILARES. MANIFESTACIONES CLÍNICAS: SEVERO ENTURBIAMIENTO CORNEAL + DEGENERACIÓN PIGMENTACIÓN DE LA RETINA INCAPACIDAD VISUAL. DEFORMACIÓN EN MANOS, FACIE GROTESCA, BOCA ABIERTA. ENTUMECIMIENTO ARTICULAR. POSIBLE DISTURBIO SIQUIÁTRICO. ALTERACIONES VÁLVULA AÓRTICA (ESTENOSIS O REGURGITACIÓN) INTELIGENCIA NORMAL. SNC NEURONAS CORTICALES NORMALES. MPS LIGERAMENTE AUMENTADOS RESPECTO A MPS IH. LESIONES EN HÍGADO Y BAZO.")
13
ALTERACIONES EN RIÑÓN, ARTERIAS Y NÓDULOS LINFÁTICOS IDEM MPS I H
DIAGNÓSTICO: MEDICIÓN URINARIA DE MPS MARCADOS MEDICIÓN DE LA ACTIVIDAD ENZIMÁTICA CROMATOGRAFÍA DE MPS URINARIOS DIFERENCIACIÓN DE MPS IH Y MPS IS POR CURSO CLÍNICO DE AMBAS. TRATAMIENTO: PROCEDIMIENTOS QUIRÚRGICOS CORRECCIÓN DEL GLAUCOMA Y DE LA ALTERACIÓN EN VÁLVULA AÓRTICA. MPS I H/S CARACTERÍSTICAS: COMPUESTO HURLER – SCHEIE DEFICIENCIA: ALFA- L- IDURONIDASA TIPO HS MANIFESTACIONES CLÍNICAS: ES MÁS SEVERA QUE MPS IS, PERO MÁS LEVE QUE LA MPS IH FRECUENCIA 1/

14
Grado De Afectación Del SNC Grado De Afectación Del Tejido Óseo
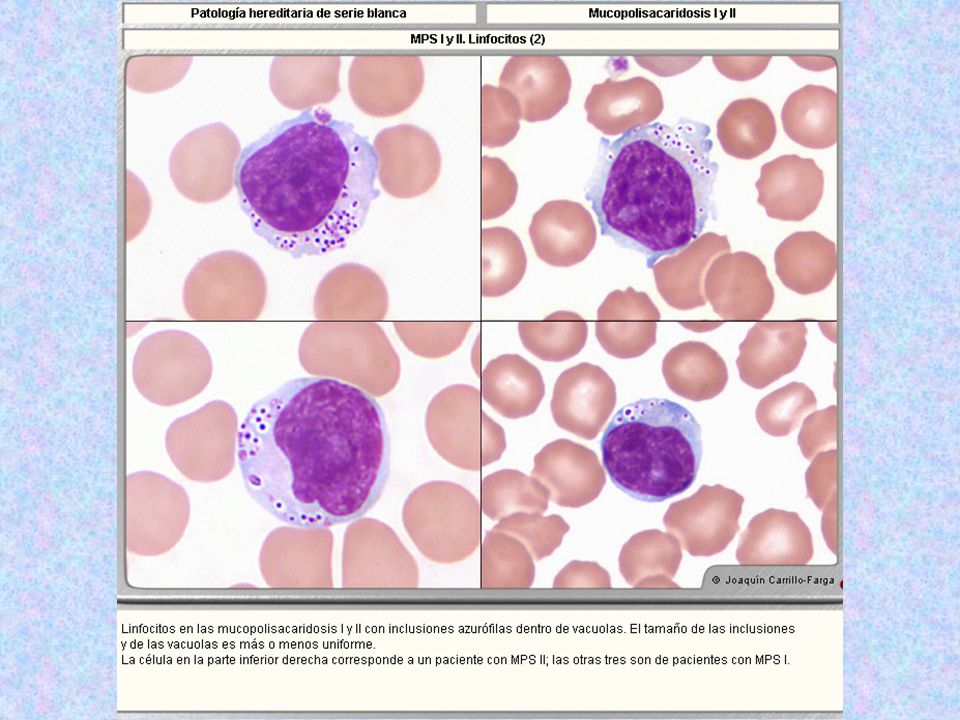
Tipo Nombre Enzima Deficiente Grado De Afectación Del SNC Grado De Afectación Del Tejido Óseo Herencia Sustrato Acumulado Rasgos Característicos Rasgos Morfológicos MPS I H MPS I S Hurler Scheie Alfa-L-Iduronidasa ++++ - + AR “ Sulfato de Dermatán y Sulfato de Heparán (menos sulfatados ) Hurler: enanismo, jorobados; opacidad corneal +++ Generalmente mueren durante la niñez. Scheie: altura normal; mano en garra; articulaciones con poca motilidad; opacidad corneal +. 5-20% de los linfocitos (Linf.B ) con gránulos azurófilos dentro de vacuolas claras: En Hunter: ++; Scheie: +.Leucocitos segmentados con granulaciones muy finas distribuidas homogéneamente.
 Hurler: enanismo, jorobados; opacidad corneal +++ Generalmente mueren durante la niñez. Scheie: altura normal; mano en garra; articulaciones con poca motilidad; opacidad corneal % de los linfocitos (Linf.B ) con gránulos azurófilos dentro de vacuolas claras: En Hunter: ++; Scheie: +.Leucocitos segmentados con granulaciones muy finas distribuidas homogéneamente.")
18
MPS II O SÍNDROME DE HUNTER
DEFICIENCIA DE IDURONIDATO SULFATASA SE ACUMULA ÁC. URÓNICO SE ALTERAN DERMATÁN Y HEPARÁN SULFATO MUTACIONES INSERCIONES (1-14BP, EXÓN 3 Y 6) SPLICING DEFECTUOSO (ELIMINACIÓN DEL EXÓN 3) PUEDE COMPARTIR COMPONENTES GENÉTICOS, (MECANISMOS REGULATORIOS) CON SULFATASAS CODIFICADAS POR GENES AUTOSÓMICOS (ASOCIACIÓN A SULFATIDOSIS) GENÉTICA: LIGADA A X LOS HETEROCIGOTAS SON FENOTÍPICAMENTE NORMALES (EFECTO CORRECTOR DE CÉLULAS NORMALES) MANIFESTACIONES CLÍNICAS: 2 FORMAS LEVE Y SEVERA ENTUMECIMIENTO ARTICULAR ENANISMO Y FACIE GROTESCA DETERIORO MENTAL PROGRESIVO LESIONES NODULARES EN LA PIEL (TORAX Y BRAZOS) PÉRDIDA PROGRESIVA DE LA VISIÓN: RETINITIS PIGMENTOSA, OPACIDAD CORNEAL CON LIGERO ENTURBIAMIENTO
 SPLICING DEFECTUOSO (ELIMINACIÓN DEL EXÓN 3) PUEDE COMPARTIR COMPONENTES GENÉTICOS, (MECANISMOS REGULATORIOS) CON SULFATASAS CODIFICADAS POR GENES AUTOSÓMICOS (ASOCIACIÓN A SULFATIDOSIS) GENÉTICA: LIGADA A X. LOS HETEROCIGOTAS SON FENOTÍPICAMENTE NORMALES (EFECTO CORRECTOR DE CÉLULAS NORMALES) MANIFESTACIONES CLÍNICAS: 2 FORMAS LEVE Y SEVERA. ENTUMECIMIENTO ARTICULAR. ENANISMO Y FACIE GROTESCA. DETERIORO MENTAL PROGRESIVO. LESIONES NODULARES EN LA PIEL (TORAX Y BRAZOS) PÉRDIDA PROGRESIVA DE LA VISIÓN: RETINITIS PIGMENTOSA, OPACIDAD CORNEAL CON LIGERO ENTURBIAMIENTO.")
19
FORMA SEVERA HIDROCEFALIA, DIARREA (SNA INTESTINAL), ADULTOS: COLORACIÓN DE LA PIEL PÓMULOS ROJIZOS APARIENCIA PLETÓRICA VOZ RONCA (OBSTRUCCIÓN RESPIRATORIATRAQUEOTOMÍA) CAUSAS DE MUERTE COMBINACIÓN VALVULAR, MIOCARDIAL E ISQUEMIA MAS DETERIORO NEUROLÓGICO DIAGNÓSTICO: DIFICIL DE DIFERENCIAR DE LAS MPS III (SF B) MUCOPOLISACARIDURIA: SE EXCRETAN EN = % DERMATÁN Y HEPARÁN TEST DE ACUMULACIÓN DE 35S-MPS EN CULTIVO DE FIBRAOBLASTOS MEDICIÓN DE LA AE EN SUERO Y CÉLULAS TEST DEL CETILPIRIDINIUM MPS URINARIOS DIAGNÓSTICO PRENATAL CULTIVO DE CÉLULAS DE LÍQUIDO ANMIÓTICO Y CUANTIFICACIÓN DE LA ACUMULACIÓN DE 35S-MPS MEDICIÓN DE AE EN CÉLULAS LÍQ. ANMIÓTICO TRATAMIENTO: INFUSIÓN DE PLASMA Y LINFOCITOS TRANSPLANTE DE MÉDULA ÓSEA TERAPIA ENZIMÁTICA TERAPIA GÉNICA
 CAUSAS DE MUERTE COMBINACIÓN VALVULAR, MIOCARDIAL E ISQUEMIA. MAS DETERIORO NEUROLÓGICO. DIAGNÓSTICO: DIFICIL DE DIFERENCIAR DE LAS MPS III (SF B) MUCOPOLISACARIDURIA: SE EXCRETAN EN = % DERMATÁN Y HEPARÁN. TEST DE ACUMULACIÓN DE 35S-MPS EN CULTIVO DE FIBRAOBLASTOS. MEDICIÓN DE LA AE EN SUERO Y CÉLULAS. TEST DEL CETILPIRIDINIUM. MPS URINARIOS. DIAGNÓSTICO PRENATAL CULTIVO DE CÉLULAS DE LÍQUIDO ANMIÓTICO Y CUANTIFICACIÓN DE LA ACUMULACIÓN DE 35S-MPS. MEDICIÓN DE AE EN CÉLULAS LÍQ. ANMIÓTICO. TRATAMIENTO: INFUSIÓN DE PLASMA Y LINFOCITOS. TRANSPLANTE DE MÉDULA ÓSEA. TERAPIA ENZIMÁTICA. TERAPIA GÉNICA.")
20
Grado De Afectación Del SNC Grado De Afectación Del Tejido Óseo
Tipo Nombre Enzima Deficiente Grado De Afectación Del SNC Grado De Afectación Del Tejido Óseo Herencia Sustrato Acumulado Rasgos Característicos Rasgos Morfológicos MPS II Hunter Iduronato-Sulfatasa a +++ Afectación menos grave sin retraso psicomotor; Afectación más grave con retraso psicomotor severo. +++ Ligada al X Sulfato de Dermatán y Sulfato de Heparán Mano en garra; fascie tosca; hipertricosis; en formas graves, retraso mental. Afectación de las válvulas cardíacas ( porque se alteran las funciones de los fibroblastos ) 5-20% de los linfocitos (Linf.B ) con gránulos azurófilos dentro de vacuolas claras +.Leucocitos segmentados con granulaciones muy finas distribuidas homogéneamente
 5-20% de los linfocitos (Linf.B ) con gránulos azurófilos dentro de vacuolas claras +.Leucocitos segmentados con granulaciones muy finas distribuidas homogéneamente.")
22
MUCOPLISACARIDOSIS III O SÍNDROME DE SANFILIPPO
DEFECTO: AUSENCIA DE LAS SIGUIENTES ENZIMAS A- HEPARAN-N-SULFATASA (HEPARAN SULFAMIDASA) B- N-AC-a - GLUCOSAMINIDASA C- N-AC-TRANSFERASA D- SULFATASA (RESIDUOS DE N-AC-GLUM-6-SULFATO) LAS CUATRO ANOMALÍAS SON CLÍNICAMENTE INDISTINGUIBLES HERENCIA AR GAG: HEPARÁN SULFATO MANIFESTACIONES CLÍNICAS: SEVERO Y PROGRESIVO RETARDO MENTAL (LENGUAJE CAPACIDAD DEL HABLA) ALTERACIONES SOMÁTICAS LEVES HEPATOMEGALIA LEVE ENANISMO LEVE ENTUMECIMIENTO ARTICULAR (< SEVERO QUE MPS I Y II) MPS A, B, C, D NO DIFERENCIABLES HISTOLÓGICAMENTE VACUOLIZACIÓN CÉLULAS HEPÁTICAS, CÉLULAS DE KUPFFER, TÚBULOS RENALES, NÓDULOS RENALES, CONDROCITOS
 B- N-AC-a - GLUCOSAMINIDASA. C- N-AC-TRANSFERASA. D- SULFATASA (RESIDUOS DE N-AC-GLUM-6-SULFATO) LAS CUATRO ANOMALÍAS SON CLÍNICAMENTE INDISTINGUIBLES. HERENCIA AR. GAG: HEPARÁN SULFATO. MANIFESTACIONES CLÍNICAS: SEVERO Y PROGRESIVO RETARDO MENTAL (LENGUAJE CAPACIDAD DEL HABLA) ALTERACIONES SOMÁTICAS LEVES. HEPATOMEGALIA LEVE. ENANISMO LEVE. ENTUMECIMIENTO ARTICULAR (< SEVERO QUE MPS I Y II) MPS A, B, C, D NO DIFERENCIABLES HISTOLÓGICAMENTE. VACUOLIZACIÓN CÉLULAS HEPÁTICAS, CÉLULAS DE KUPFFER, TÚBULOS RENALES, NÓDULOS RENALES, CONDROCITOS.")
23
AL MICROSCOPIO ELECTRÓNICO SE OBSERVAN CUERPOS DE CEBRA INCLUSIONES GRÁNULOS VACUOLARES EN SNC
MUERTE 20 AÑOS MPS III B ALTERACIONES EN LA VÁLVULA MITRAL DIAGNÓSTICO PRESUNTIVO AUMENTO DE EXCRESIÓN URINARIA DE HEPARÁN SULFATO TEST DE AZUL DE TOLUIDINA (-) ACUMULACION DE 35-S- MPS EN CULTIVO FIBROBLASTOS DETERMINACIÓN DE N-AC-a- D- GLUCOSAMINIDASA MPS III B: POSIBLE DIAGNÓSTICO PRENATAL DETERMINACIÓN DE N-AC-a- D- GLUCOSAMINIDASA EN HETEROCIGOTAS TERAPIA: SANGRE TOTAL, SUERO, LEUCOCITOS TRANSPLANTE DE MÉDULA ÓSEA
 ACUMULACION DE 35-S- MPS EN CULTIVO FIBROBLASTOS. DETERMINACIÓN DE N-AC-a- D- GLUCOSAMINIDASA. MPS III B: POSIBLE DIAGNÓSTICO PRENATAL DETERMINACIÓN DE N-AC-a- D- GLUCOSAMINIDASA EN HETEROCIGOTAS. TERAPIA: SANGRE TOTAL, SUERO, LEUCOCITOS. TRANSPLANTE DE MÉDULA ÓSEA.")
25
Grado De Afectación Del SNC Grado De Afectación Del Tejido Óseo
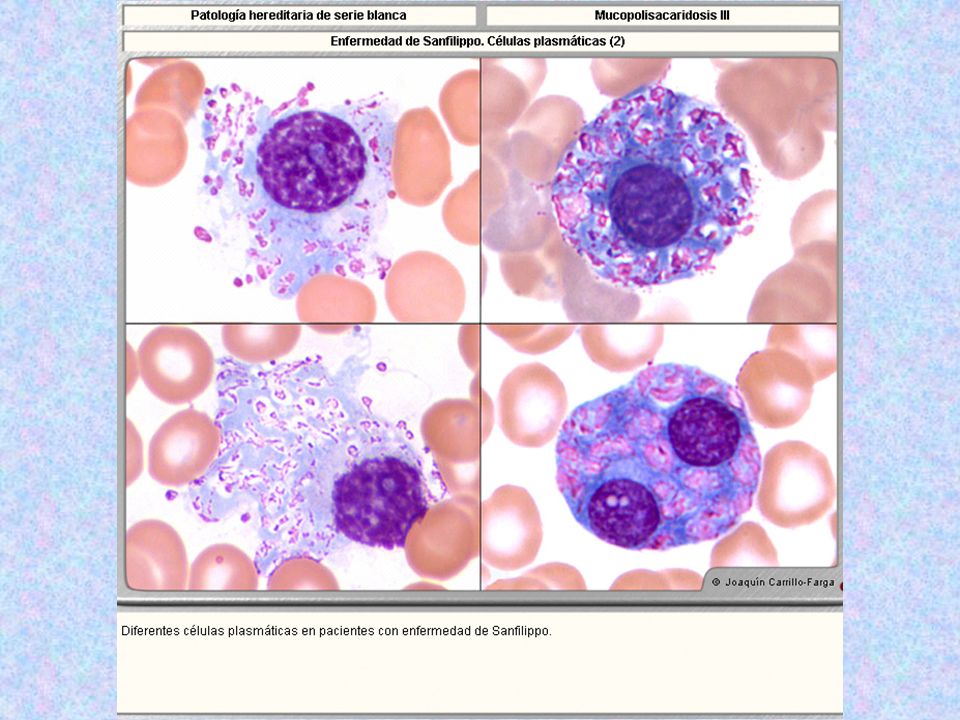
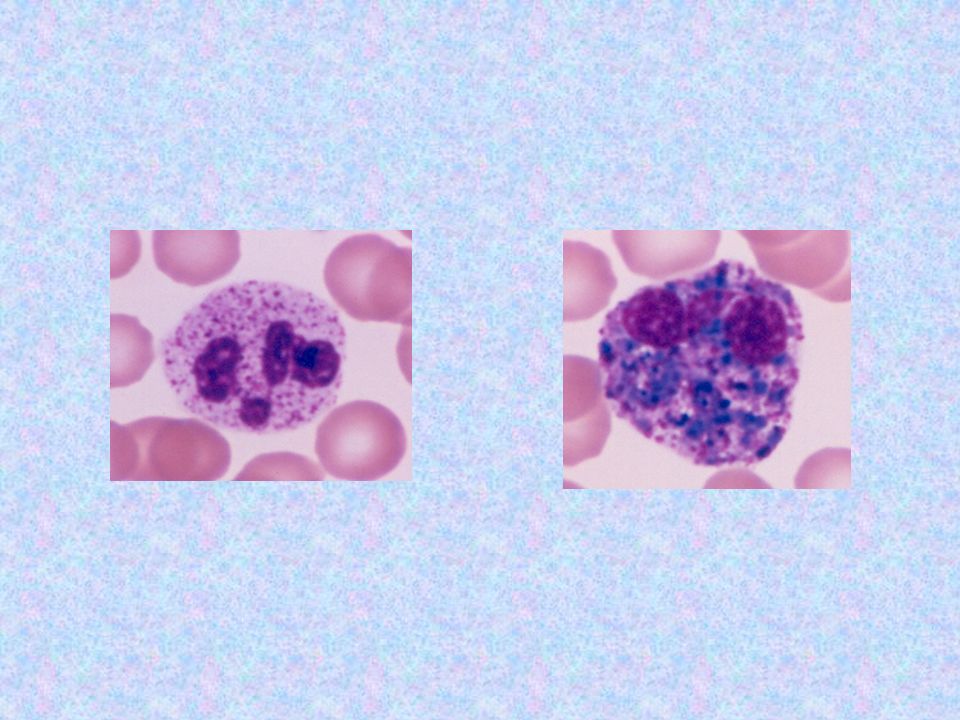
Tipo Nombre Enzima Deficiente Grado De Afectación Del SNC Grado De Afectación Del Tejido Óseo Herencia Sustrato Acumulado Rasgos Característicos Rasgos Morfológicos MPS III SanFilippo Heparán-sulfatasa +++ ++ AR Sulfato de Heparán Mano en garra; se desarrollan bien hasta la adolescencia, en la que empiezan a retroceder iniciando con demencia y progresivo deterioro mental e hipertricosis. 5-20% de los linfocitos (Linf.B ) con gránulos azurófilos dentro de vacuolas claras +. M.O.: células plasmáticas anormales el 100%, con vacuolas con un gránulo azurófilo en su interior; a veces los gránulos se aplastan y se ven como letras chinas.
 con gránulos azurófilos dentro de vacuolas claras +. M.O.: células plasmáticas anormales el 100%, con vacuolas con un gránulo azurófilo en su interior; a veces los gránulos se aplastan y se ven como letras chinas.")
28
MUCOPOLISACARIDOSIS IV O SÍNDROME DE MORQUIO QUERATÁN SULFATURIA
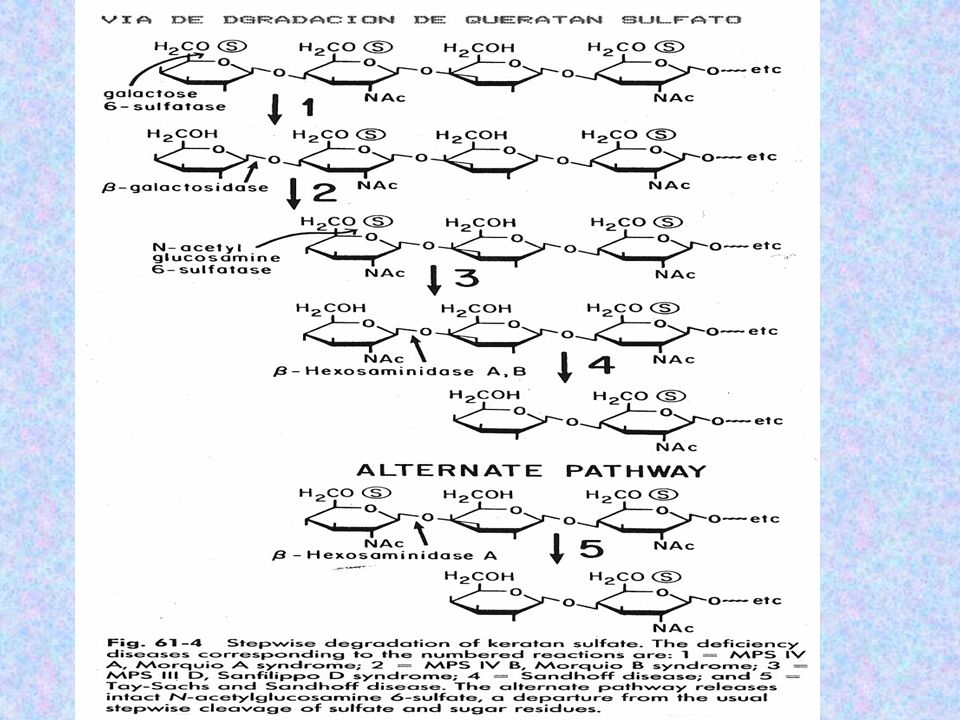
DEFECTO: DEFICIENCIA EN LA DEGRADACIÓN DE QUERATÁN SULFATO (CARTÍLAGO) SULFATASA CUYOS SUSTRATOS SERÍAN GALACTOSA -6-SULFATO Y N-AC-GLUCOSAMINA-6-SULFATO HERENCIA: AR CLASIFICACIÓN: TIPO A: DEFICIENCIA DE N-AC-GAL-6-SULFATASA MUTACIÓN EXÓN 1, DOBLE DELECCIÓN TIPO B: b- GALACTOSIDASA (HIDRÓLISIS QUERATÁN SULFATO) MUTACIÓN: SUSTITUCIONES, DUPLICACIONES, INSERCIONES Y MUTACIONES EN SITIOS DE SPLICING
 SULFATASA CUYOS SUSTRATOS SERÍAN GALACTOSA -6-SULFATO Y N-AC-GLUCOSAMINA-6-SULFATO. HERENCIA: AR. CLASIFICACIÓN: TIPO A: DEFICIENCIA DE N-AC-GAL-6-SULFATASA MUTACIÓN EXÓN 1, DOBLE DELECCIÓN. TIPO B: b- GALACTOSIDASA (HIDRÓLISIS QUERATÁN SULFATO) MUTACIÓN: SUSTITUCIONES, DUPLICACIONES, INSERCIONES Y MUTACIONES EN SITIOS DE SPLICING.")
29
MANIFESTACIONES CLÍNICAS:
EFECTOS SOBRE EL CORDÓN ESPINAL (SN) RASGOS FACIALES GROSEROS (BOCA GRANDE Y DIENTES ESPARCIDOS) SEVERAS DEFORMACIONES ÓSEAS RODILLAS MUY JUNTAS, TRONCO CORTO, PROMINENTE ESTERNÓN, CUELLO CORTO Y CABEZA SOBRE LOS HOMBROS) SE DETIENE EL CRECIMIENTO 6 A 7 AÑOS ARTICULACIONES SUELTAS SORDERA LEVE ENTURBIAMIENTO CORNEAL PARÁLISIS RESPIRATORIA ALTERACIONES CARDÍACAS HISTOLOGÍA: MO Y ME SE OBSERVAN GRANDES VACUOLAS EN CEREBRO Y CARTÍLAGOS DIAGNÓSTICO: QUERATÁN SULFATO URINARIO CROMATOGRAFÍA EN PLACA FINA DETERMINACIÓN ENZIMÁTICA VNTR SOUTHERN BLOT TRATAMIENTO: CIRUGÍA ORTOPÉDICA
 RASGOS FACIALES GROSEROS (BOCA GRANDE Y DIENTES ESPARCIDOS) SEVERAS DEFORMACIONES ÓSEAS RODILLAS MUY JUNTAS, TRONCO CORTO, PROMINENTE ESTERNÓN, CUELLO CORTO Y CABEZA SOBRE LOS HOMBROS) SE DETIENE EL CRECIMIENTO 6 A 7 AÑOS. ARTICULACIONES SUELTAS. SORDERA. LEVE ENTURBIAMIENTO CORNEAL. PARÁLISIS RESPIRATORIA. ALTERACIONES CARDÍACAS. HISTOLOGÍA: MO Y ME SE OBSERVAN GRANDES VACUOLAS EN CEREBRO Y CARTÍLAGOS. DIAGNÓSTICO: QUERATÁN SULFATO URINARIO CROMATOGRAFÍA EN PLACA FINA. DETERMINACIÓN ENZIMÁTICA. VNTR. SOUTHERN BLOT. TRATAMIENTO: CIRUGÍA ORTOPÉDICA.")
30
Galactosa-6-sulfatasa
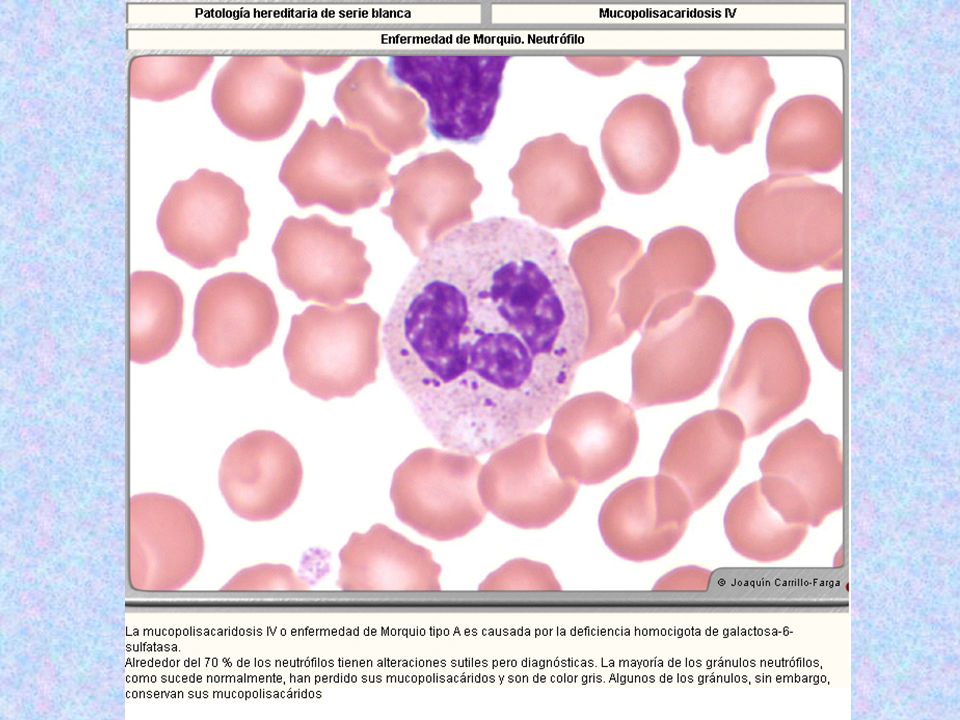
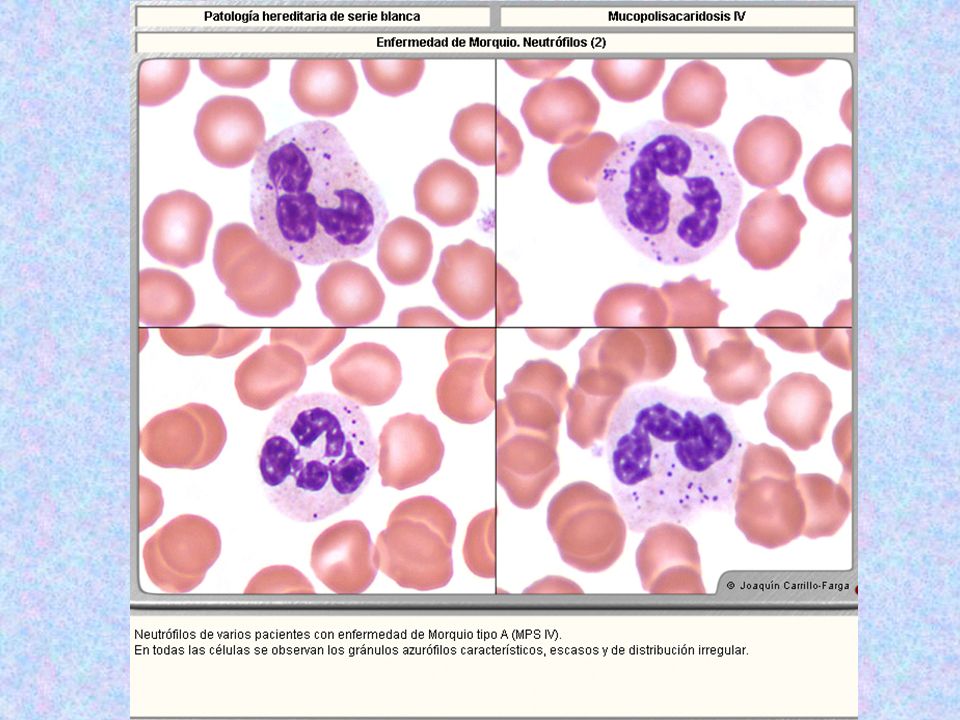
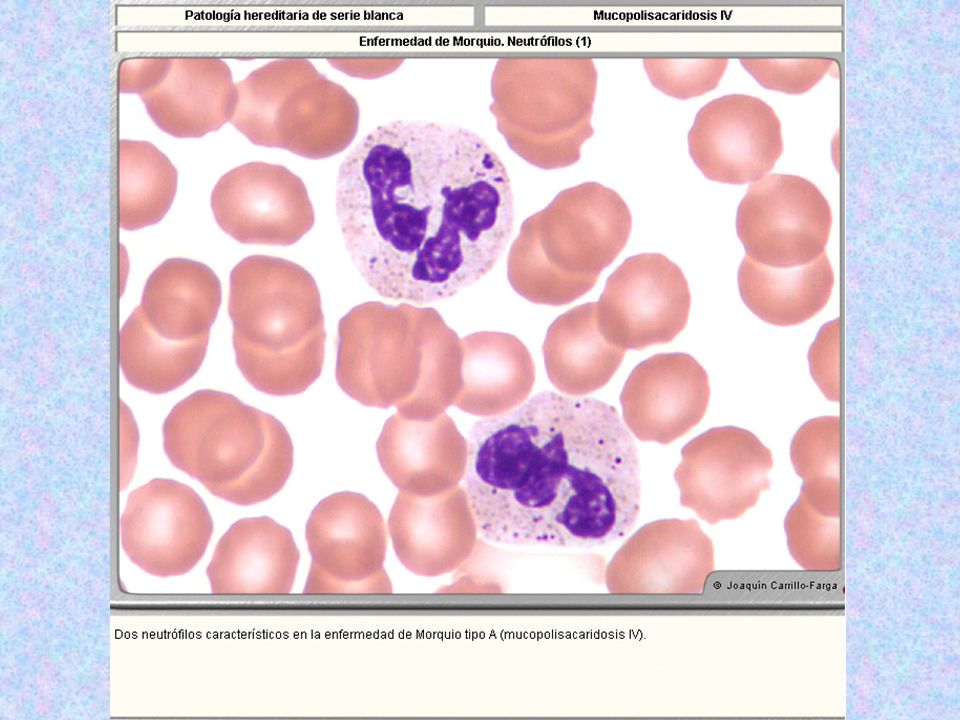
Tipo Nombre Enzima Deficiente Grado De Afectación Del SNC Grado De Afectación Del Tejido Óseo Herencia Sustrato Acumulado Rasgos Característicos Rasgos Morfológicos MPS IV * Morquio Tipo A Galactosa-6-sulfatasa - ++++ AR Sulfato de Keratán Sin cuello; cara tosca; miembros deformes; pecho en quilla; joroba; sin alteraciones intelectuales. Opacidad corneal. Linfocitos con gránulos azurófilos dentro de vacuolas claras. Gránulos azurófilos acumulados en grupos distribuidos irregularmente, en el 80% de los neutrófilos.

34
MUCOPOLISACARIDOSIS V
ANTERIORMENTE ASIGNADA A SÍNDROME DE SCHEIE, HOY VACANTE MUCOPOLISACARIDOSIS VI O SÍNDROME DE MAROTEAUX LAMY DEFICIENCIA: N-AC-GALACTOSAMINA-4-SULFATASA O ARILSULFATASA B ACTÚA SOBRE SUSTRATO SINTÉTICO HERENCIA: AR MANIFESTACIONES CLÍNICAS: 3 FORMAS 1- SEVERA 2- INTERMEDIA 3- LEVE 1- SEVERA - 2-3 AÑOS, RETARDO EVIDENTE EN EL DESARROLLO -RESTRICCIÓN MOVIMIENTO ARTICULAR (CADERA Y FÉMUR) -PROTRUSIÓN ESTERNAL ANTERIOR -ENTURBIAMIENTO CORNEAL -ANORMALIDADES CARDÍACAS -HIDROCEFALIA
 -PROTRUSIÓN ESTERNAL ANTERIOR. -ENTURBIAMIENTO CORNEAL. -ANORMALIDADES CARDÍACAS. -HIDROCEFALIA.")
35
2- LEVE -ESTATURA BAJA DENSO ENTURBIAMIENTO CORNEAL HERNIAS INGUINALES ESTENOSIS AÓRTICAS MICROSCOPÍA INCLUSIONES LISOSOMALES IGUALES QUE EN MPS I Y II EN CÉLULAS DE KUPFFER (TB. EN CONJUNTIVA, CÓRNEAS, Y PIEL) DIAGNÓSTICO: DETERMINACIÓN URINARIA DE DERMATÁN SULFATO MEDICIÓN DE ARIL SULFATASA B DIAGNÓSTICO PRENATAL EN LÍQUIDO ANMIÓTICO
 DIAGNÓSTICO: DETERMINACIÓN URINARIA DE DERMATÁN SULFATO. MEDICIÓN DE ARIL SULFATASA B. DIAGNÓSTICO PRENATAL EN LÍQUIDO ANMIÓTICO.")
36
Grado De Afectación Del SNC Grado De Afectación Del Tejido Óseo
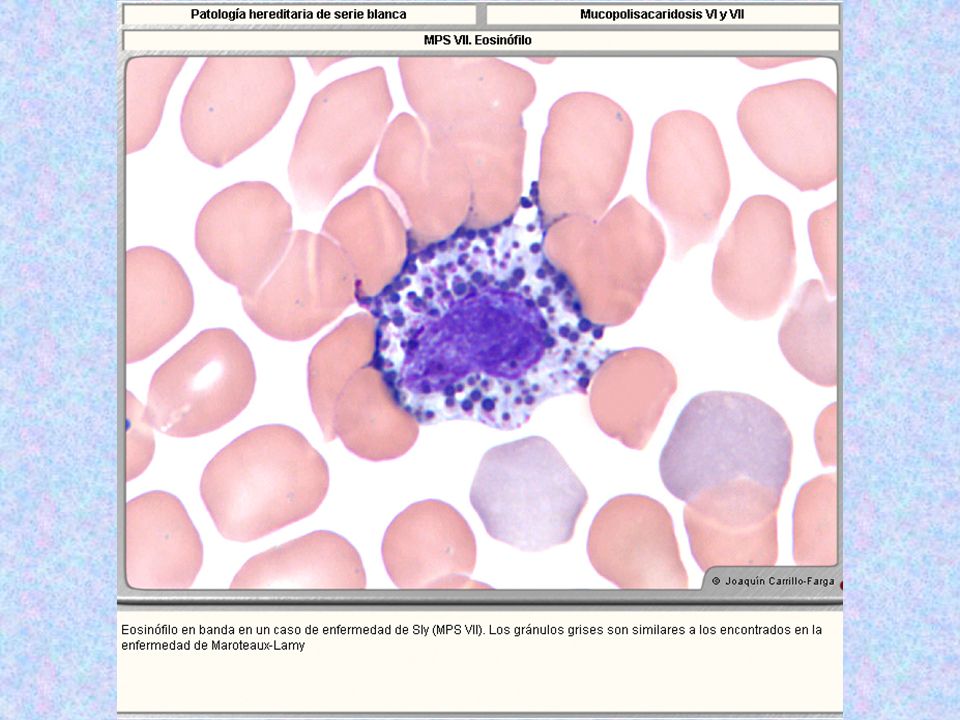
Tipo Nombre Enzima Deficiente Grado De Afectación Del SNC Grado De Afectación Del Tejido Óseo Herencia Sustrato Acumulado Rasgos Característicos Rasgos Morfológicos MPS VI* Maroteaux-Lamy Aril-Sulfatasa-B ( la mayor parte de los MPS son degradados por esta enzima). - ++++ AR Sulfato de Dermatán Sulfato de Condroitín Inteligencia normal; pecho en quilla; cara tosca. Presencia de muchos gránulos azurófilos; indistinguibles de las granulaciones tóxicas, pero sin datos clin. ni de lab. propios de las mismas. Eosinófilos con gránulos grises por la mezcla de MPS no degradados y prot. Catiónicas (alteraciones patognomónicas ). Monocitos con gránulos azurófilos más marcados. Menos del 10% de los linfocitos idénticos a los de MPS I y II.
 AR. Sulfato de Dermatán. Sulfato de Condroitín. Inteligencia normal; pecho en quilla; cara tosca. Presencia de muchos gránulos azurófilos; indistinguibles de las granulaciones tóxicas, pero sin datos clin. ni de lab. propios de las mismas. Eosinófilos con gránulos grises por la mezcla de MPS no degradados y prot. Catiónicas (alteraciones patognomónicas ). Monocitos con gránulos azurófilos más marcados. Menos del 10% de los linfocitos idénticos a los de MPS I y II.")
37
Maroteaux-lamy

39
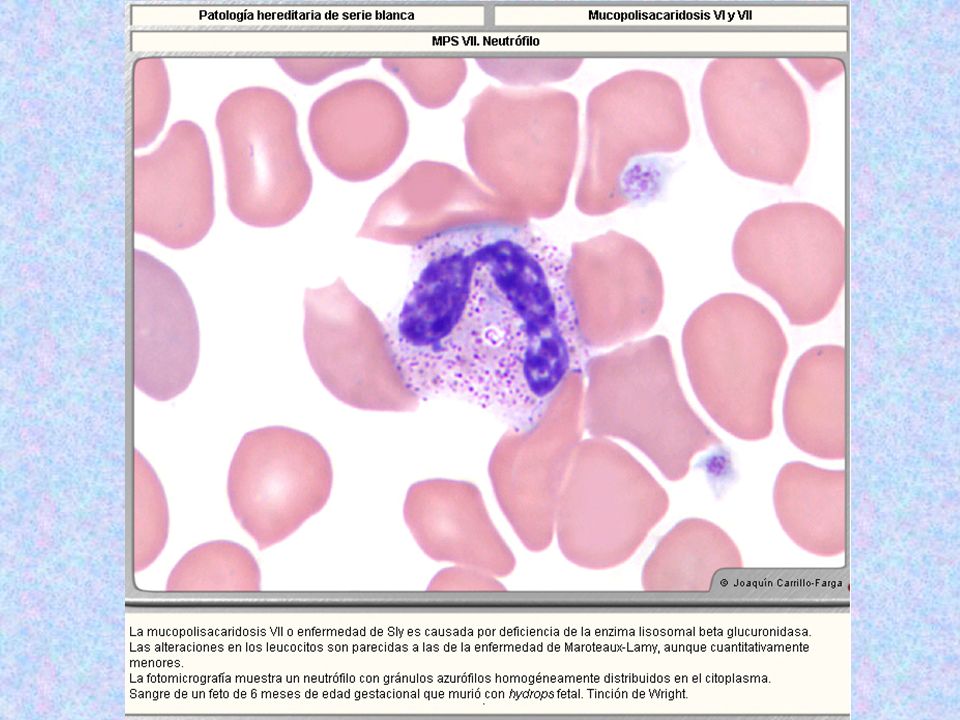
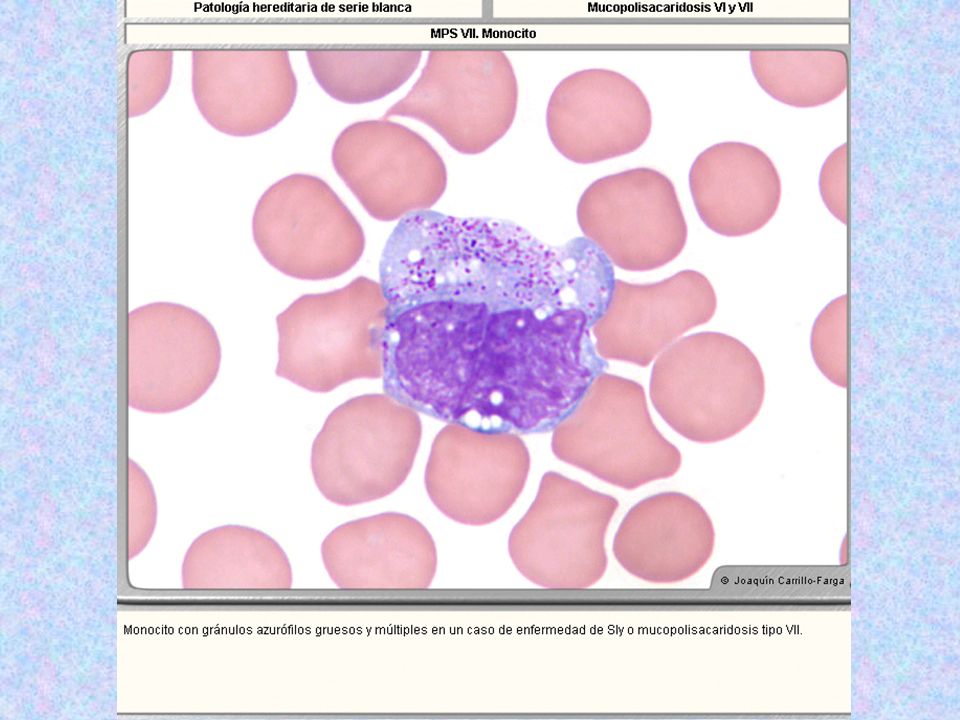
MUCOPOLISACARIDOSIS VII O DE SLY
DEFICIENCIA: b- GLUCURONIDASA BLOQUEO DE LA DEGRADACIÓN DE DERMATÁN SULFATO (FIBROBLASTOS, LEUCOCITOS) Y DE HEPARÁN SULFATO. LA ENZIMA SE ENCUENTRA INMUNOLÓGICAMENTE INTACTA HERENCIA: AR MANIFESTACIONES CLÍNICAS: -FACIE INUSUAL -DEFORMACIÓN TORÁCICO- LUMBAR -HEPATOESPLENOMEGALIA -HERNIAS UMBILICALES -DETERIORO FÍSICO Y MENTAL HISTOLOGÍA: GRUESAS GRANULACIONES EN GRANULOCITOS Y MÉDULA ÓSEA DIAGNÓSTICO: DETERMINACIÓN DE LA ACTIVIDAD DE b- GLUCURONIDASA EN FIBROBLASTOS, LEUCOCITOS Y SUERO TERAPIA: - TERAPIA GÉNICA EXPERIMENTAL EN RATONES (VECTOR)
 Y DE HEPARÁN SULFATO. LA ENZIMA SE ENCUENTRA INMUNOLÓGICAMENTE INTACTA. HERENCIA: AR. MANIFESTACIONES CLÍNICAS: -FACIE INUSUAL. -DEFORMACIÓN TORÁCICO- LUMBAR. -HEPATOESPLENOMEGALIA. -HERNIAS UMBILICALES. -DETERIORO FÍSICO Y MENTAL. HISTOLOGÍA: GRUESAS GRANULACIONES EN GRANULOCITOS Y MÉDULA ÓSEA. DIAGNÓSTICO: DETERMINACIÓN DE LA ACTIVIDAD DE b- GLUCURONIDASA EN FIBROBLASTOS, LEUCOCITOS Y SUERO. TERAPIA: - TERAPIA GÉNICA EXPERIMENTAL EN RATONES (VECTOR)")
40
Grado De Afectación Del SNC Grado De Afectación Del Tejido Óseo
Tipo Nombre Enzima Deficiente Grado De Afectación Del SNC Grado De Afectación Del Tejido Óseo Herencia Sustrato Acumulado Rasgos Característicos Rasgos Morfológicos MPS VII* Sly Beta-glucuronidasa. Secundaria a la Aril-sulfatasa B, y papel muy similar al de ésta en la deg.de los MPS de neutrófilos y eosinófilos. Muy Variable, desde muerte in utero por hydrops fetalis, hasta pacientes adultos normales. + AR Sulfato de Heparán Datos clínicos muy similares a los de Maroteaux Lamy, pero en menor grado. Muchos pacientes mueren de Hydrops fetalis. Neutrófilos con gránulos azurófilos no tan abundantes como en Maroteaux-Lamy:: mismo aspecto que Maroteaux-Lamy, pero en menor grado; menos gránulos azurófilos, menos acúmulo de MPS.

47
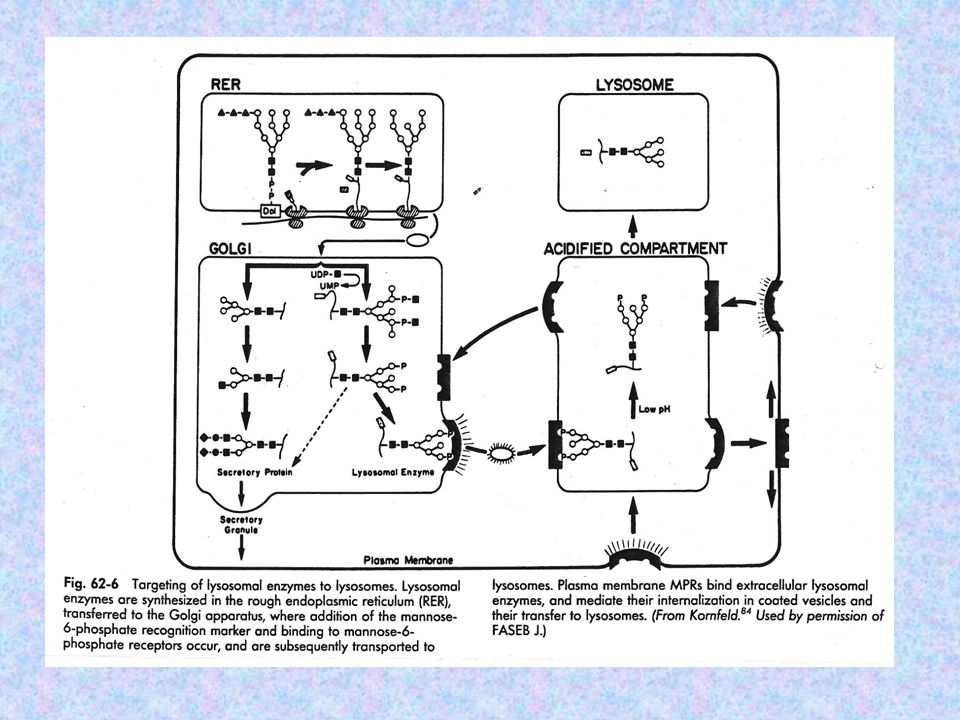
MUCOLIPIDOSIS DEFICIENCIA DE HIDROLASAS LISOSOMALES (TEJIDOS Y FIBROBLASTOS), NECESARIAS PARA EL CATABOLISMO DE MPS SE OBSERVA EL AUMENTO DE LOS NIVELES DE HIDROLASAS LISOSOMALES EN FLUÍDOS CORPORALES FALLAN LAS MODIFICACIONES POSTRADUCCIONALES DE LAS ENZIMAS (RESIDUOS MANOSA-6-FOSFATO) LAS ENZIMAS QUE SE ENCUENTRAN ELEVADAS SON: a-L- IDURONIDASA IDURONATO SULFATASA b- GLUCURONIDASA N-AC-b-HEXOSAMINIDASA ARIL SULFATASA a- MANOSIDASA a- L-FUCOSIDASA FUNCIÓN CATABOLISMO DE MPS, GLICOLÍPIDOS Y GLICOPROTEÍNAS LAS ENZIMAS QUE SE ENCUENTRAN NORMALES SON: b- GLUCOSIDASA FOSFATASA ÁCIDA
 LAS ENZIMAS QUE SE ENCUENTRAN ELEVADAS SON: a-L- IDURONIDASA. IDURONATO SULFATASA. b- GLUCURONIDASA. N-AC-b-HEXOSAMINIDASA. ARIL SULFATASA. a- MANOSIDASA. a- L-FUCOSIDASA. FUNCIÓN CATABOLISMO DE MPS, GLICOLÍPIDOS Y GLICOPROTEÍNAS. LAS ENZIMAS QUE SE ENCUENTRAN NORMALES SON: b- GLUCOSIDASA. FOSFATASA ÁCIDA.")
48
CLASIFICACIÓN MUCOLIPIDOSIS TIPO II TIPO III
MUCOLIPIDOSIS TIPO II O DESÓRDEN DE LAS CÉLULAS I DEFICIENCIA: LAS ENZIMAS SINTETIZADAS POR LOS FIBROBLASTOS NO SON FOSFORILADAS HERENCIA: AR MANIFESTACIONES CLÍNICAS: TEMPRANAS COMPLICACIONES CARDIORRESPIRATORIAS SEVERAS SIMILARES A MPS I H TRASTORNOS CARDÍACOS RECIEN NACIDO DISLOCACIÓN CONGÉNITA (CADERA, TORAX, HERNIAS) RETARDO PSICOMOTOR PROGRESIVO MUERTE 5 O 6 AÑOS DIAGNÓSTICO: DETERMINACIÓN DE LA ACTIVIDAD DE LAS ENZIMAS LISOSOMALES EN SUERO O PLASMA PRENATAL EN CULTIVO DE CÉLULAS DE LÍQUIDO ANMIÓTICO TERAPIA NO HAY
 RETARDO PSICOMOTOR PROGRESIVO. MUERTE 5 O 6 AÑOS. DIAGNÓSTICO: DETERMINACIÓN DE LA ACTIVIDAD DE LAS ENZIMAS LISOSOMALES. EN SUERO O PLASMA. PRENATAL EN CULTIVO DE CÉLULAS DE LÍQUIDO ANMIÓTICO. TERAPIA NO HAY.")
49
MUCOLIPIDOSIS TIPO III
DEFICIENCIA HIDROLASAS NO FOSFORILADAS O CON BAJO RENDIMIENTO EN LA FOSFORILACIÓN ALTERACIÓN DEL CAMINO ENZIMÁTICO QUE FOSFORILA LOS RESIDUOS DE MANOSA-6-FOSFATO HERENCIA: AR MANIFESTACIONES CLÍNICAS: SIMILARES (HISTOLÓGICAMENTE) A ML TIPO II DIAGNÓSTICO: IDEM ML TIPO II TERAPIA:
 A ML TIPO II. DIAGNÓSTICO: IDEM ML TIPO II. TERAPIA:")
50
Grado De Afectación Del SNC Grado De Afectación Del Tejido Óseo
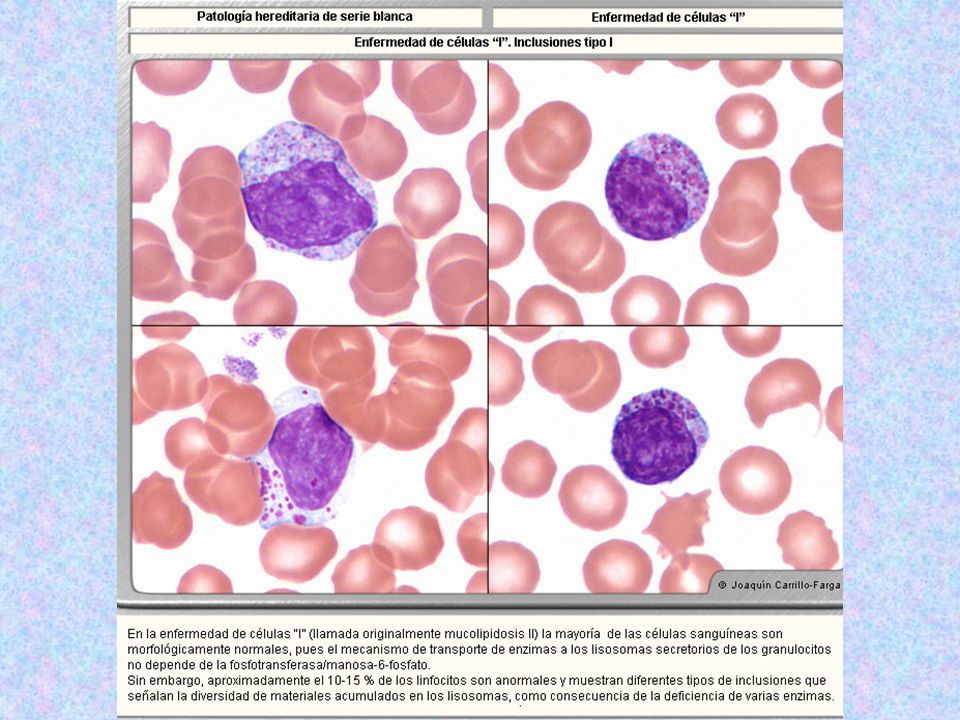
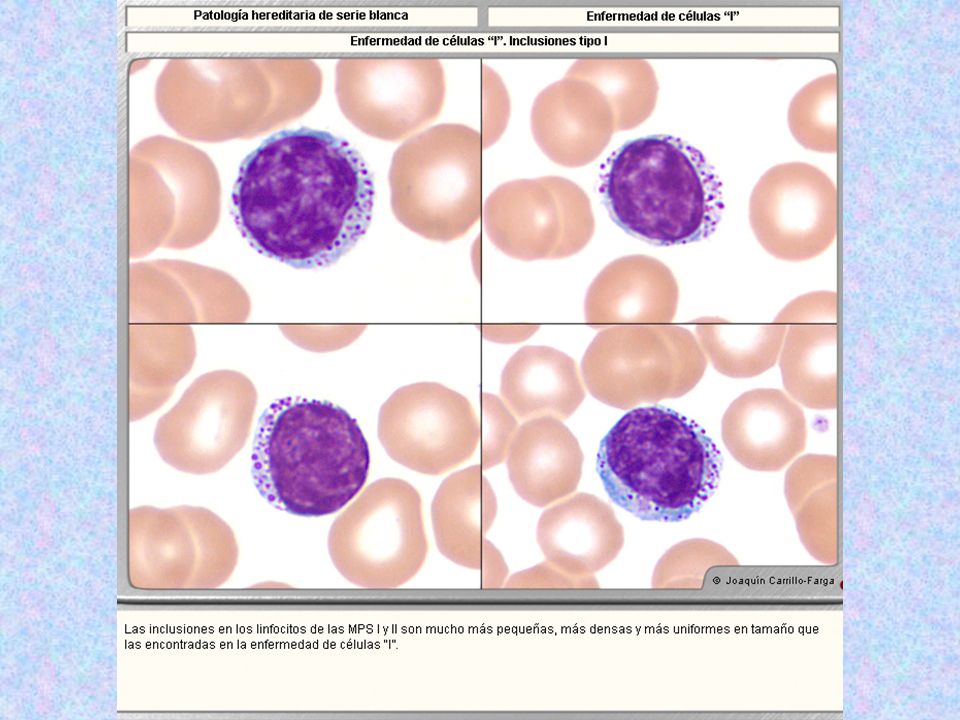
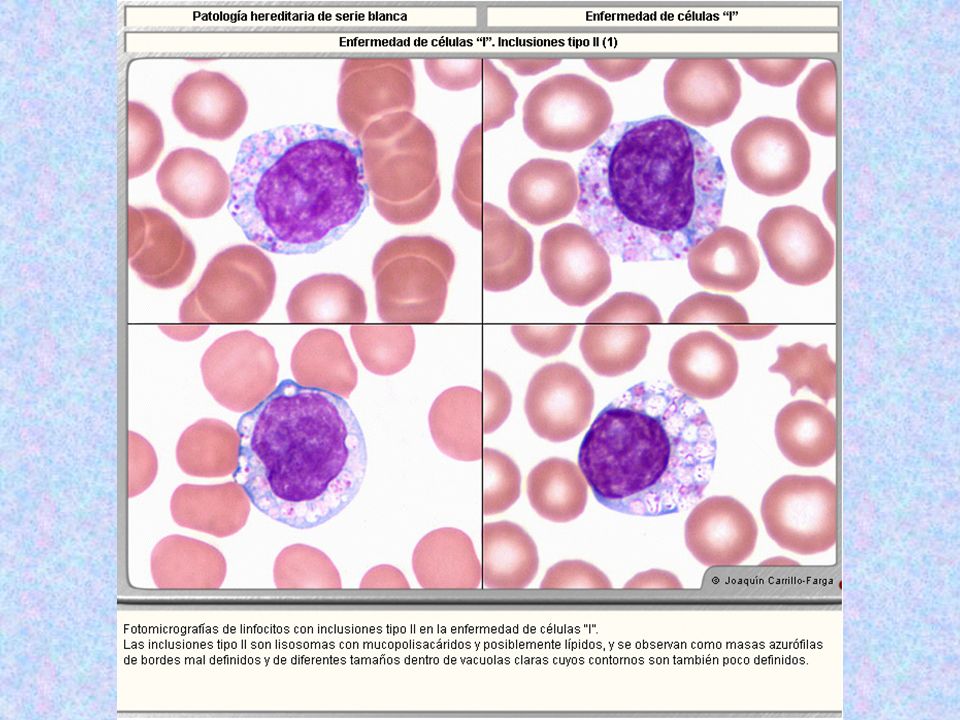
Tipo Nombre Enzima Deficiente Grado De Afectación Del SNC Grado De Afectación Del Tejido Óseo Herencia Sustrato Acumulado Rasgos Característicos Rasgos Morfológicos Muco lipido sis II Enferme dad de Células I ( antes Hurler neonatal ) Fosfotransferas Las enzimas ( proteínas lisosomales, no secretorias ) en el Golgi no son marcadas con Manosa, y en lugar de unirse a los receptores de Manosa, salen al exterior, como si fueran proteínas de secreción. ( ahí no dañan, pues no funcionan debido al pH inapropiado ).El daño se produce por el acúmulo de muchas sustancias ( MPS y lípidos) + Mezcla de MPS y de Lípidos Hipertrofia de encías, cara tosca, hipopigmentación del pelo( las enzimas no llegan bien al melanoso ma, aunque es un tipo de lisosoma secretorio, pero en ellos también es importante el mecanismo de la manosa-6-fosfato. No hay alteraciones en neutrófilos, eosinófilos, basófilos, monocitos, ya que estas células no utilizan el mecanismo de la manosa-6-fosfato. Sólo el linfocito B sí lo utiliza, y serán las únicas células de S.P. afectadas, con los siguientes 3 tipos celulares en el 10% de todos los linfocitos: Inclusiones tipo 1:Lisosomas llenos de MPS, de aspecto de gránulos grandes, sólidos, no se ven dentro de vacuolas. Inclusiones Tipo 2.( más comunes ) Inclusiones de varios tamaños, de bordes mal definidos, situados dentro de vacuolas de diferentes tamaños, que constituyen lisosomas anómalos. Inclusiones tipo 3.( raras ). Vacuolas vacías. También existen acantocitos, por la alteración de lípidos plasmáticos.
 Fosfotransferas. Las enzimas. ( proteínas lisosomales, no secretorias ) en el Golgi no son marcadas con Manosa, y en lugar de unirse a los receptores de Manosa, salen al exterior, como si fueran proteínas de secreción. ( ahí no dañan, pues no funcionan debido al pH inapropiado ).El daño se produce por el acúmulo de muchas sustancias. ( MPS y lípidos) + Mezcla de MPS y de Lípidos. Hipertrofia de encías, cara tosca, hipopigmentación del pelo( las enzimas no llegan bien al melanoso ma, aunque es un tipo de lisosoma secretorio, pero en ellos también es importante el mecanismo de la manosa-6-fosfato. No hay alteraciones en neutrófilos, eosinófilos, basófilos, monocitos, ya que estas células no utilizan el mecanismo de la manosa-6-fosfato. Sólo el linfocito B sí lo utiliza, y serán las únicas células de S.P. afectadas, con los siguientes 3 tipos celulares en el 10% de todos los linfocitos: Inclusiones tipo 1:Lisosomas llenos de MPS, de aspecto de gránulos grandes, sólidos, no se ven dentro de vacuolas. Inclusiones Tipo 2.( más comunes ) Inclusiones de varios tamaños, de bordes mal definidos, situados dentro de vacuolas de diferentes tamaños, que constituyen lisosomas anómalos. Inclusiones tipo 3.( raras ). Vacuolas vacías. También existen acantocitos, por la alteración de lípidos plasmáticos.")
Presentaciones similares










>")
















