Descargar la presentación
La descarga está en progreso. Por favor, espere

1
Caso clinico Ateneo Interhospitalario
Clinica Medica Hosp. J.M. Cullen Dr. Sandiano Dra. Folmer Schimpf

2
Antecedentes Femenina, 31 años Tabaquista 18 paquetes/año
Padre fallece por cáncer de cabeza y cuello Litiasis ureteral y múltiples episodios de pielonefritis Septiembre 2011: hipertensión arterial. Se medica con enalapril 10mg c/12hs

3
Motivo de consulta Dolor lumbar de 3 meses de evolución bilateral a predominio derecho de intensidad 9-10, de tipo continuo, que cede parcialmente con analgésicos, que no se exacerba con los movimientos y no cede con el reposo Cefalea de 6 meses de evolución, bitemporal, de tipo pulsátil, intermitente, que cede con AINES

4
Examen fisico TA:120/60 mmHg FC:80lpm FR:16rpm T: 36.2°C
Cabeza y cuello: nódulo tiroideo en lóbulo izquierdo CV: R1 y R2 NF. Sin soplo. Sin edemas. Sin ingurgitación yugular. Pulsos periféricos conservados y simétricos Ap. Resp.: buena entrada bilateral de aire, sin ruidos agregados Ap. Dig.: blando, depresible, indoloro, RHA+, timpánico. Sin visceromegalia

5
Examen fisico Ap. Genitourinario: puñopercusión renal bilateral negativa. Mamas sin nódulo palpable, simétricas, movilidad conservada Sin adenopatias axilares, cervicales, inguinales SOMA: dolor a la palpación superficial de fosa lumbar. Lasegue (-). Percusión de columna (-)
. Percusión de columna (-)")
6
Examen fisico Piel y faneras: Distribución ginecoide de vello pubiano. Sin hipertricosis. Linea de implantación de pelo s/p. Sin giba, ni estrias rojovinosas. Nevo de 10x7cm en región de cresta ilíaca izquierda (congénito)
")
9
Laboratorio GB (63/29/8) HB 12.7 g/L Hto 39.8% plaquetas VES 5mm/h GPT 43.2 U/l GOT 16.5 FAL 187 BT y BD Suero no icterico Proteinas totales 6.67 Albumina 3.94 g/l Urea 0.30 mg/dl Glicemia 0.72 g/l Creatinina 0.73 mg/dl K 4.4 mEq/L Na 139 mEq/L

10
Diagnósticos presuntivos
Métodos complementarios

11
Oftalmologia: papilas simétricas, bordes netos, coloracion normal
Oftalmologia: papilas simétricas, bordes netos, coloracion normal. Macula libre. Fondo de ojo normal. Ecocardiograma bidimensional: normal

12
Orina de 24 hs Volumen remitido 3.000 ml Proteinuria 24hs 395 mg/24hs Na U 156 mEq/24hs K U 27.1 mEq/24hs

13
Resumen Paciente Joven Hipertensión arterial
Sin antecedente familiar de hipertensión arterial Cefalea intermitente Dolor lumbar Nódulo tiroideo Sin daño de órgano blanco HTA secundaria?

14
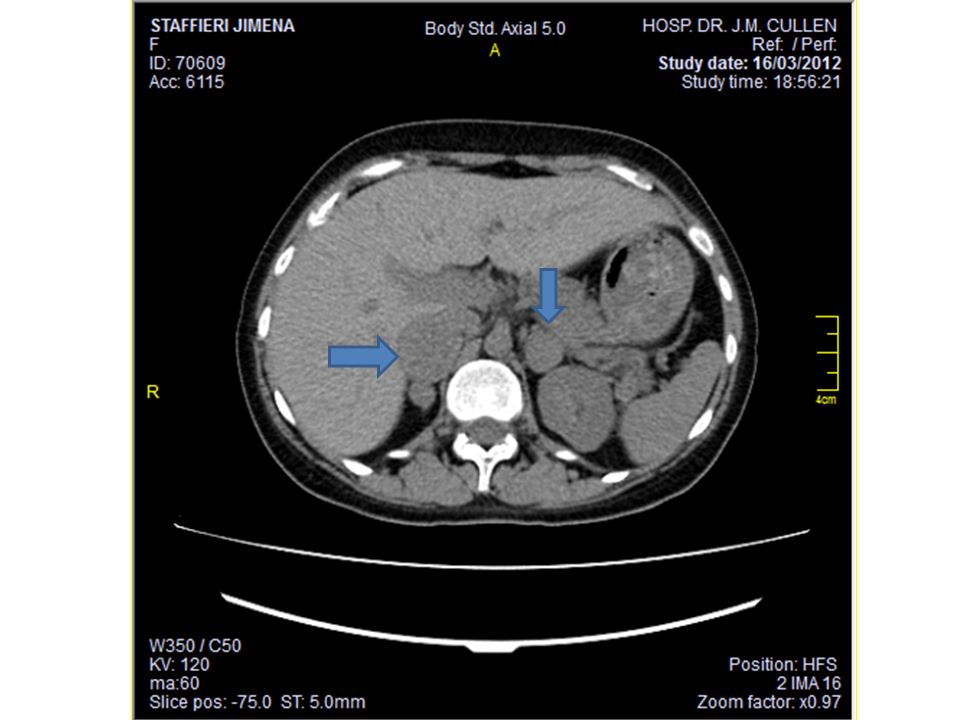
TAC de abdomen Lesiones en ambas glandulas suprarrenales
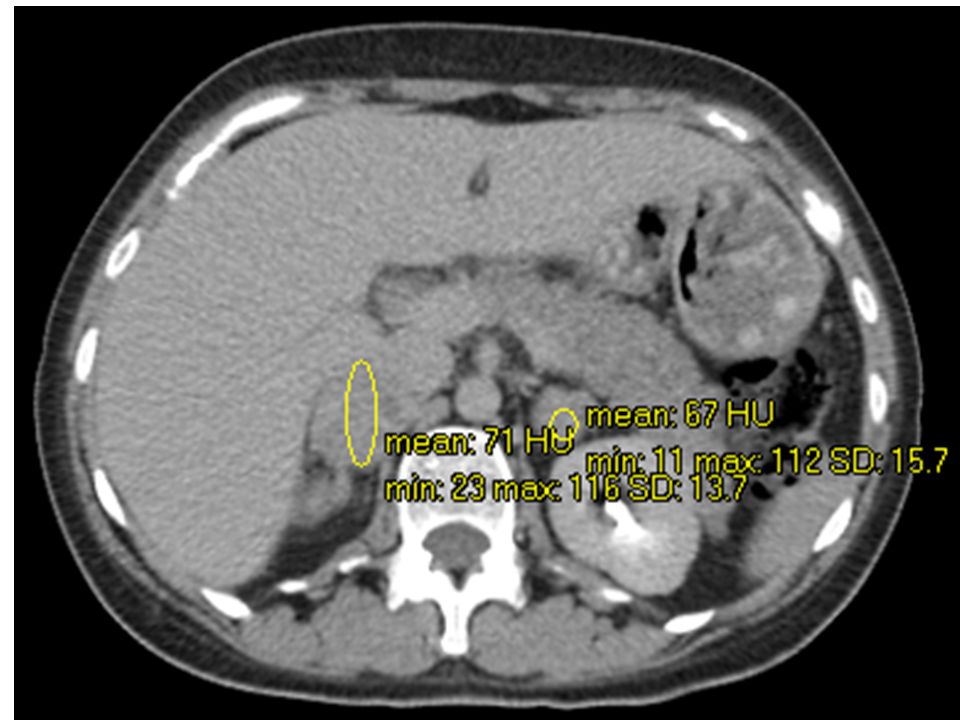
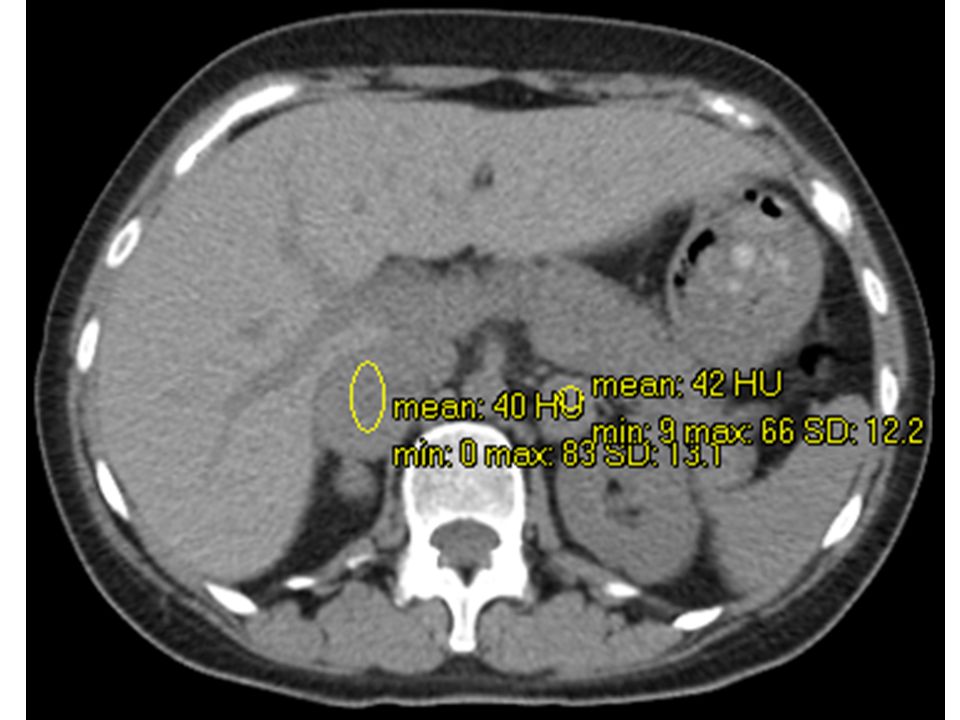
Suprarrenal derecha: imagen nodular con diámetro mayor de 36mm, áreas hipodensas que pueden representar necrosis Suprarrenal izquierda: diámetro mayor de 28mm, con hallazgos tisulares similares Higado, bazo, via biliar, pancreas y riñones s/p GSD: UH. Washout lento (GSI similar)
")
18
Diagnósticos presuntivos
Métodos complementarios

19
Laboratorio Aldosterona 190 pg/ml (10-160 pg/ml)
DHEA sulfato 2.8 ug/dl (60-400ug/dl) Renina plasmatica 32.4 pg/ml ( pg/ml) Metanefrinas urinarias 398 ug/24hs ( ug/24hs) Normetanefrina urinarias 410 ug/24hs ( ug/24hs) Dopamina (metodo HPLC) 28 pg/ml (<50pg/ml) Adrenalina plasmatica 32 pg/ml ( pg/ml) Cortisol libre urinario 78 ug/24 hs ( ug/24hs)
 Renina plasmatica 32.4 pg/ml ( pg/ml) Metanefrinas urinarias 398 ug/24hs ( ug/24hs) Normetanefrina urinarias 410 ug/24hs ( ug/24hs) Dopamina (metodo HPLC) 28 pg/ml (<50pg/ml) Adrenalina plasmatica 32 pg/ml ( pg/ml) Cortisol libre urinario 78 ug/24 hs ( ug/24hs)")
20
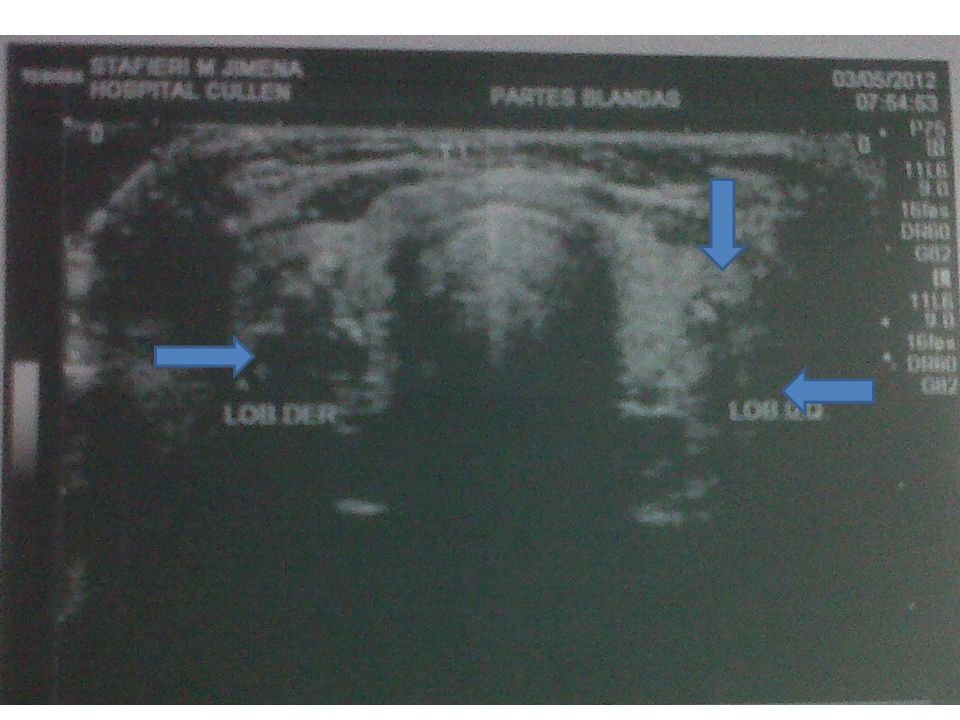
Ecografia Glandula tiroidea de contornos regulares Lobulo derecho
en 1/3 medio formación hipoecoica heterogénea, de contornos irregulares, con calcificaciones, de 13x15, con circulación periférica al doppler color Diámetro 64x15x16 Lobulo izquierdo en 1/3 medio, dos formaciones de aspecto quístico anecoicas de contornos irregulares, con contenido ecogénico en su interior, de 9 y 7mm Diámetro 47x15x13 Itsmo heterogéneo

22
Laboratorio T4 libre 1.08 ug/dl TSH 0.86 mlU/L Calcemia 11 mg/dl
Parathormona pg/ml (10-55 pg/ml) FSH 2.04 mUI/ml LH 1.39 CEA 11.8 ng/ml Prolactina ng/ml Calcitonina 16 pg/ml (≤10pg/ml)
 FSH 2.04 mUI/ml. LH CEA 11.8 ng/ml. Prolactina ng/ml. Calcitonina 16 pg/ml (≤10pg/ml)")
23
PAAF de lóbulo tiroideo derecho
Anatomía patológica informa 3 celulas atípicas, extendido no concluyente

24
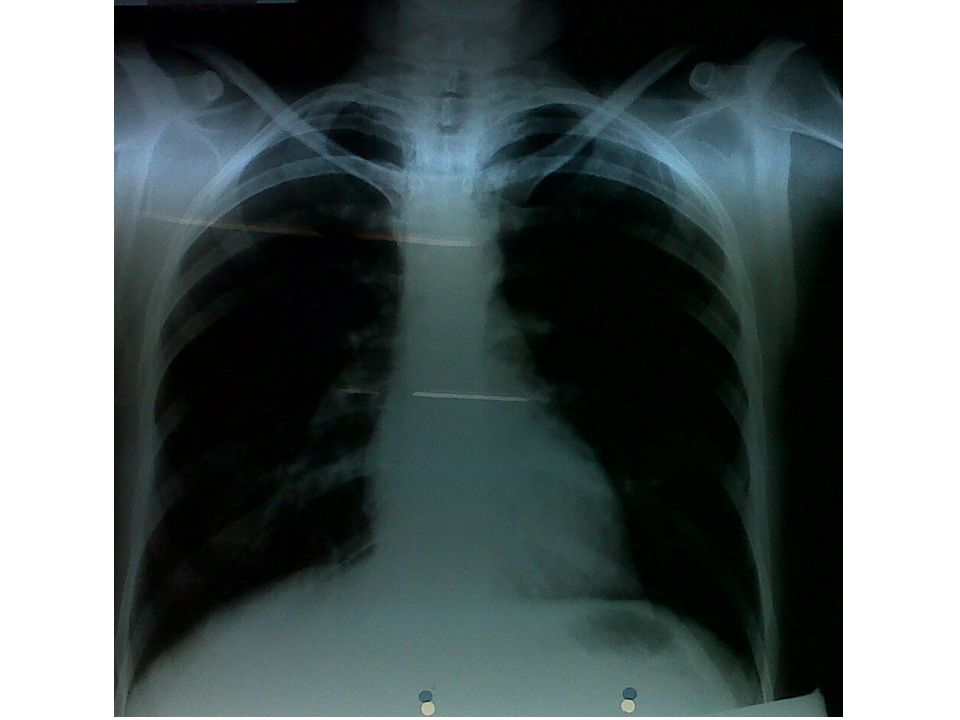
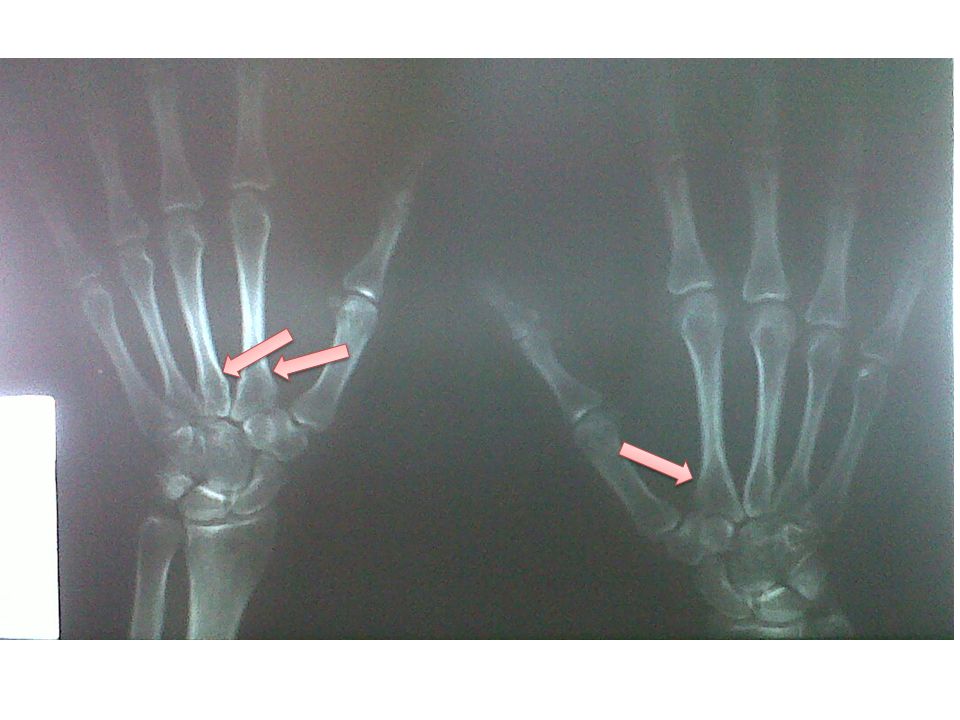
Radiografias

30
Centellograma con sestamibi
Área de hiperfijación nodular precoz en proyección del lóbulo izquierdo superior, que persiste a las 3-4hs en imágenes planares y spect, compatible con nódulo paratiroideo hiperfuncionante como primer diagnóstico diferencial

31
RMN de craneo Adenohipofisis de morfología e intensidad de señal normal Sin imágenes nodulares dominantes Sin areas de realce patológico luego de la inyección de contraste EV El tallo hipofisiario es central Senos cavernos simetricos Estructuras vasculares de la región selar sin alteraciones Neurohipofisis con señal habitual en T1 y T2en T2 en la totalidad del encéfalo no se indentifican efectos de masa, ni colecciones intraparenquimatosas

32
Resumen HTA secundaria Sospecha de feocromocitoma bilateral
Nódulo tiroideo, malignidad?

33
Se realiza suprarrenalectomía bilateral, total, por vía laparoscopica
Inicia terapia de reemplazo hormonal (hidrotisona y fludrocortisona)
")
34
ANATOMIA GLANDULAS SUPRARRENALES
SUPRARRENAL DERECHA: formacion ovoidea que mide 6x4, 5x3cm. Al corte, parenquima glandular reemplazado por formacion de aspecto nodular, de coloracion rosado, palido, con areas congestivas hemorragicas, rodea a dicha formacion parenquima remanente de coloracion naranja-dorado. SUPRARRENAL IZQUIERDA: que junto a tejido adiposo mide 5x4x1cm, sobre la superficie externa se observa una formacion nodular de similares caracteristicas a lo descripto anteriormente, mide 2.5x2x2cm. EXAMEN MICROSCOPICO: en patron alveolar, trabecular predominante y areas solidas (focales, escasa), las celulas son intermedias o poligonas, con grande citoplasma y granulos. Se ven nucleos hipercromicos y vacuolas intracitoplasmaticas. No se observan mitosis. Se reconoce infiltracion capsular focal y permeacion vascular venular, en dos sitios. DIAGNOSTICO: feocromocitoma bilateral H-E 40X H-E 40X Derecha Izquierda H-E 4X H-E 4X CELULAS INTERMEDIAS CON ABUNDANTE CITOPLASMA CON GRANULOS Y NUCLEOS HIPERCCROMICOS
, las celulas son intermedias o poligonas, con grande citoplasma y granulos. Se ven nucleos hipercromicos y vacuolas intracitoplasmaticas. No se observan mitosis. Se reconoce infiltracion capsular focal y permeacion vascular venular, en dos sitios. DIAGNOSTICO: feocromocitoma bilateral. H-E 40X. H-E 40X. Derecha. Izquierda. H-E 4X. H-E 4X. CELULAS INTERMEDIAS CON ABUNDANTE CITOPLASMA CON GRANULOS Y NUCLEOS HIPERCCROMICOS.")
35
Se realiza tiroidectomía total y paratiroidectomía parcial
Inicia terapia de reemplazo hormonal (levotiroxina)
")
36
ANATOMIA PATOLOGICA TIROIDEA
Izquierda Derecha H-E 40X H-E 4X H-E 40X ROJO CONGO 4X H-E 4X ROJO CONGO 4X BIRREFRINGENCIA DE RC A LUZ POLARIZADA EXAMEN MACROSCOPICO: pieza de tiroidectomia total. Lobulo derecho mide 5x3x2.5 cm, al corte y a nivel del 1/3 medio del lobulo, formacion nodular que mide 1.3x1x1 cm de tonalidad blanquecina y aspecto heterogeneo. Lobulo izquierdo de 4.5x2.5x1.5cm, al corte, sobre 1/3 medio se observan 2 formaciones nodulares, miden 0.5cm de diametro mayor cada uno, bien circunscripto, distan entre si 0.4cm. Ambas presentan caracteristicas similares a las descriptas en lesion de lobulo derecho. Itsmo mide 2.4x1.3x0.7cm con caracteristicas habituales. Material remitido como glandula paratiroides: formacion nodular que mide 2x0.5cm de coloracion blanco-amarronada y superficie lisa. Al corte tonalidad amarronada homogenea DIAGNOSTICO INTRAOPERATORIO: POSITIVO PARA CELULAS NEOPLASICAS Diagnostico patologico Carcinoma medular de tiroides, multifocal, bilateral Capsula tiroidea libre de compromiso neoplasico CELULAS PLASMOCITOIDES TRABECULAS DE MATERIAL SIMIL AMILOIDE

37
IHQ 40X 40X Cromogranina Calcitonina

38
Factor de transcripcion tiroideo N 1 (TTF-1)
40X Factor de transcripcion nuclear: factor de transcripcion tiroideo n1 Factor de transcripcion tiroideo N 1 (TTF-1)
")
39
GLANDULA PARATIROIDES
H-E 10X H-E 40X HIPERPLASIA A EXPENSAS DE CELULAS CLARAS

40
NEM 2A Diagnostico Feocromocitoma bilateral
Carcinoma medular de tiroides Hiperparatiroidismo ESTUDIO MOLECULAR: se amplificaron 2 fragmentos de 186pb y 296pb al exon 10 y 11 del protooncogen RET (10q11.2). Se detecto mutacion Cys634Arg heterocigota (TGC→CGC) compatible con NEM 2A
. Se detecto mutacion Cys634Arg heterocigota (TGC→CGC) compatible con. NEM 2A.")
41
Neoplasias endocrinas múltiples
Se caracterizan por la presencia de tumores que involucran dos o más glándulas endocrinas en un mismo paciente. Su prevalencia se estima entre 20 y 200 casos por 1 millón de habitantes. Tienen una expresión variable y los síntomas muchas veces son leves. Son trastornos raros, de herencia autonómica dominante. Los síndromes clásicos son los NEM, tipo 1 y 2 (A, B y CMTF).
.")
42
NEM TIPO 1 Es la asociación de tumores ubicados en las paratiroides, la hipófisis y el páncreas. Trastorno autosomico dominante que aparece por mutaciones inactivadoras del gen de la NEM1 (MEN1), localizado en el cromosoma 11q13, que codifica la una proteína supresora tumoral llamada menina. Clínica muy variable, en cuanto al numero de sistemas orgánicos afectados, la edad de inicio de los tumores y de los síntomas, tanto dentro de las familias como entre ellas. La mayoría se diagnostica en la adolescencia o inicio de la adultez. La manifestación más frecuente es el Hiperparatiroidismo (95%). Los Tumores Pancreáticos - Duodenales , en 2do lugar. (gastrinomas (50%) e insulinomas (33%) y, menos frecuentes, el glucagonoma, tumores productores de VIP o de PP (polipéptido pancreático). El tumor hipofisario se presenta en el 65% de las NEM 1. Pueden secretar PRL, GH, ACTH y el resto pareciera ser no funcionante. La clínica dependerá del tamaño del tumor y de las hormonas secretadas.
, localizado en el cromosoma 11q13, que codifica la una proteína supresora tumoral llamada menina. Clínica muy variable, en cuanto al numero de sistemas orgánicos afectados, la edad de inicio de los tumores y de los síntomas, tanto dentro de las familias como entre ellas. La mayoría se diagnostica en la adolescencia o inicio de la adultez. La manifestación más frecuente es el Hiperparatiroidismo (95%). Los Tumores Pancreáticos - Duodenales , en 2do lugar. (gastrinomas (50%) e insulinomas (33%) y, menos frecuentes, el glucagonoma, tumores productores de VIP o de PP (polipéptido pancreático). El tumor hipofisario se presenta en el 65% de las NEM 1. Pueden secretar PRL, GH, ACTH y el resto pareciera ser no funcionante. La clínica dependerá del tamaño del tumor y de las hormonas secretadas.")
43
NEM TIPO 1 Se hace un diagnóstico clínico de NEM1 en pacientes con tumores en 2/3 órganos que se afectan con mayor frecuencia (paratiroides, hipófisis y tumores pancreáticos/duodenales) y en pacientes con uno de tales tumores y un antecedente familiar de NEM1. También se debe sospechar en pacientes con : HPT primario en menores de 30 años de edad, HPTP con afectacion multiglandular, HPTP familiar, sindrome de Zollinger-Ellison y en los tumores endocrinos pancreaticos multifocales. Ex. Complementarios: La medición de calcemia y PTH, se consideran exámenes útiles y fáciles de realizar. Otros: medición de hormonas gastrointestinales y prolactina, y la exploración radiológica del abdomen y la hipófisis. Analisis Genético: sobre todo en jóvenes, permite vigilar con anticipación la aparición tumores relacionados con la NEM1. Tratamiento: Dependerá de los tumores presentes en el paciente, algunos de tto. Medico y otros tto. Quirurgico.
 y en pacientes con uno de tales tumores y un antecedente familiar de NEM1. También se debe sospechar en pacientes con : HPT primario en menores de 30 años de edad, HPTP con afectacion multiglandular, HPTP familiar, sindrome de Zollinger-Ellison y en los tumores endocrinos pancreaticos multifocales. Ex. Complementarios: La medición de calcemia y PTH, se consideran exámenes útiles y fáciles de realizar. Otros: medición de hormonas gastrointestinales y prolactina, y la exploración radiológica del abdomen y la hipófisis. Analisis Genético: sobre todo en jóvenes, permite vigilar con anticipación la aparición tumores relacionados con la NEM1. Tratamiento: Dependerá de los tumores presentes en el paciente, algunos de tto. Medico y otros tto. Quirurgico.")
44
NEM TIPO 2 Características Moleculares Las mutaciones en el proto-oncogen Ret (c-Ret), ubicado en el cromosoma 10q 11.2, son causantes de las NEM 2. El RET codifica un receptor tirosina-cinasa que funciona como transductor de la señal tras la interacción con la familia de ligandos del factor neurotrófico de origen glial. Las mutaciones producen una activación independiente del ligando y dan lugar al crecimiento y a la supervivencia de la célula. Se requiere sólo una copia del gen mutado para lograr el efecto fenotípico. La mutación del codón 634 representa el 80% de todas las mutaciones identificadas en la neoplasia endocrina múltiple tipo 2 y se expresa como un NEM 2A. La manifestación más común y característica de las NEM 2 es el riesgo muy alto de sufrir carcinoma medular de tiroides (CMT) a lo largo de la vida (95%)
, ubicado en el cromosoma 10q 11.2, son causantes de las NEM 2. El RET codifica un receptor tirosina-cinasa que funciona como transductor de la señal tras la interacción con la familia de ligandos del factor neurotrófico de origen glial. Las mutaciones producen una activación independiente del ligando y dan lugar al crecimiento y a la supervivencia de la célula. Se requiere sólo una copia del gen mutado para lograr el efecto fenotípico. La mutación del codón 634 representa el 80% de todas las mutaciones identificadas en la neoplasia endocrina múltiple tipo 2 y se expresa como un NEM 2A. La manifestación más común y característica de las NEM 2 es el riesgo muy alto de sufrir carcinoma medular de tiroides (CMT) a lo largo de la vida (95%)")
45
NEM TIPO 2 NEM 2A (80%) se caracteriza, además de CMT (100%), por la presencia de Feocromocitoma uni o bilateral (50%) e Hiperparatiroidismo (20%). La mayoría de los pacientes con NEM2 A tienen un progenitor afectado. La mutación mas frecuente tiene lugar en la cisteína en el codón 634 en el exon 11. Hay un pequeño numero de familias con NEM2 A que tienen amiloidosis de tipo liquen cutaneo pruriginoso (exantema pruriginoso en espalda) o enfermedad de Hirschsprung. NEM 2B considerada la de peor pronóstico, siendo el CMT (100%) de aparición mucho más precoz que en las otras formas, presenta Feocromocitoma en 30-50%, No hay compromiso de paratiroides A diferencia de las otras variedades, existen anormalidades esqueléticas (hábito marfanoide 97%), alteraciones oftalmológicas (prominencia corneal, engrosamiento palpebral, neuromas subconjuntivales) neuromas bucales y ganglioneuromatosis gastrointestinal (mas del 90%). Carcinoma Medular de Tiroides Familiar: Cuatro o mas casos de CMT con Ausencia demostrada de feocromocitoma e hiperparatiroidismo. El CMT en este grupo tiende a ser menos activo que en el resto de los subtipos de NEM2 y tiende a tener la edad mas avanzada de inicio.
 o enfermedad de Hirschsprung. NEM 2B. considerada la de peor pronóstico, siendo el CMT (100%) de aparición mucho más precoz que en las otras formas, presenta Feocromocitoma en 30-50%, No hay compromiso de paratiroides. A diferencia de las otras variedades, existen anormalidades esqueléticas (hábito marfanoide 97%), alteraciones oftalmológicas (prominencia corneal, engrosamiento palpebral, neuromas subconjuntivales) neuromas bucales y ganglioneuromatosis gastrointestinal (mas del 90%). Carcinoma Medular de Tiroides Familiar: Cuatro o mas casos de CMT con Ausencia demostrada de feocromocitoma e hiperparatiroidismo. El CMT en este grupo tiende a ser menos activo que en el resto de los subtipos de NEM2 y tiende a tener la edad mas avanzada de inicio.")
46
Carcinoma Medular de Tiroides
Esporádico (85%) o Familiar (15%). Representa cerca de 5 a 10% de los cánceres de tiroides. Se origina en las células parafoliculares "C", las cuales secretan calcitonina. Hay tres formas Familiares de CMT: MEN 2A, MEN 2B y CMTF. Tiene la característica de secretar Calcitonina, marcador útil para estatificación, enfermedad residual y seguimiento. Un nivel sérico basal que supere los 20 pg/ml justifica en un pte. con un nódulo tiroideo, una investigación más a fondo que descarte el CTM. Neoplasia relativamente agresiva localmente, al momento del diagnóstico un 60-80% tiene metástasis linfáticas. Se asocia a altas tasas de enfermedad persistente y recidiva, aunque los pacientes suelen vivir por largos períodos Clínica: Nódulo uni-bilateral. Adenopatías cervicales (50%), síntomas compresivos (15%) como disfagia, tos o ronquera. La secreción de calcitonina puede dar lugar a diarrea y flushing facial. Metástasis a distancia en hueso, hígado y pulmones.
 o Familiar (15%). Representa cerca de 5 a 10% de los cánceres de tiroides. Se origina en las células parafoliculares C , las cuales secretan calcitonina. Hay tres formas Familiares de CMT: MEN 2A, MEN 2B y CMTF. Tiene la característica de secretar Calcitonina, marcador útil para estatificación, enfermedad residual y seguimiento. Un nivel sérico basal que supere los 20 pg/ml justifica en un pte. con un nódulo tiroideo, una investigación más a fondo que descarte el CTM. Neoplasia relativamente agresiva localmente, al momento del diagnóstico un 60-80% tiene metástasis linfáticas. Se asocia a altas tasas de enfermedad persistente y recidiva, aunque los pacientes suelen vivir por largos períodos. Clínica: Nódulo uni-bilateral. Adenopatías cervicales (50%), síntomas compresivos (15%) como disfagia, tos o ronquera. La secreción de calcitonina puede dar lugar a diarrea y flushing facial. Metástasis a distancia en hueso, hígado y pulmones.")
47
Carcinoma Medular de Tiroides
El Diagnóstico generalmente se hace luego de una PAAF con tinción IHQ, ecografía, la medición de calcitonina sérica y CEA. Siempre se debe descartar en el preoperatorio de un CMT, el hiperparatiroidismo y el feocromocitoma como parte de un NEM. Se sugiere calcemia y metanefrinas plasmáticas/catecolaminas urinarias de 24hs. El Tratamiento es principalmente quirúrgico. Tiroidectomia Total + diseccion de ganglios linfaticos centrales +/- laterales ipsilaterales/contralaterales. RTX y QMT paliativa si enfermedad avanzada. En todos los pacientes con CMT está justificado investigar la existencia de mutaciones del gen RET, ya que es posible ofrecer consejo genético y realizar pruebas genéticas a los familiares y estratificar el riesgo.

48
Hiperparatiroidismo Primario
Incidencia del 1% en la población general, la cual se incrementa con la edad y aparece en el 20 al 30% de los pacientes con NEM2 A. Puede deberse a un solo adenoma o a una hiperplasia de todas las glándulas. La mayoría de los pacientes se encuentra asintomático y se llega incidentalmente al diagnostico ante una hipercalcemia. Clínica: Si aparecen síntomas, son similares a los del HPT esporádico: nefrolitiasis, reducción de la DMO, que produce osteopenia u osteoporosis, astenia, miopatía, enfermedad ulcerosa péptica y déficits neurocognitivos, incluidos la depresión y la alteración del sueño. El Diagnóstico se confirma con un valor alto o normal-alto de la calcemia junto con la PTH sérica elevada. El Tratamiento es Quirúrgico. Paratiroidectomía Subtotal/Total + Timectomia Transcervical (MEN1). Paratiroidectomía Subtotal/Total (MEN2) Localizacion Preoperatoria: Centellograma con Sestamibi
. Paratiroidectomía Subtotal/Total (MEN2) Localizacion Preoperatoria: Centellograma con Sestamibi.")
49
Feocromocitoma Se originan en la medula suprarrenal o celulas cromafines de la cadena ganglionar simpática (paragangliomas). Producen, almacenan y secretan catecolaminas. Se observa en 0.1% aproximadamente de los sujetos Hipertensos y en el 6,5% de los incidentalomas suprarrenales. “Tumor del 10%” (el 10% son extraadrenales; el 10% son múltiples o bilaterales; el 10% recidiva tras la cirugía; el 10% son malignos; el 10% son familiares) En los pacientes con NEM es bilateral en el 50% de los pacientes y con frecuencia es benigno. Tríada Clásica: Cefalea episódica, sudoración y taquicardia. Cerca de un 50% presenta tanto Hipertensión Paroxística, HTA esencial o incluso Normotension ( 10%). Diagnóstico : La mayor sensibilidad la tienen las metanefrinas plasmáticas libres (S 99%), seguidas de las metanefrinas urinarias fraccionadas (S 97%) (orina de 24hs). La determinación menos sensible fue la de AVM (S 64%) pero a su vez las mas especifica (E 95%).
. Producen, almacenan y secretan catecolaminas. Se observa en 0.1% aproximadamente de los sujetos Hipertensos y en el 6,5% de los incidentalomas suprarrenales. Tumor del 10% (el 10% son extraadrenales; el 10% son múltiples o bilaterales; el 10% recidiva tras la cirugía; el 10% son malignos; el 10% son familiares) En los pacientes con NEM es bilateral en el 50% de los pacientes y con frecuencia es benigno. Tríada Clásica: Cefalea episódica, sudoración y taquicardia. Cerca de un 50% presenta tanto Hipertensión Paroxística, HTA esencial o incluso Normotension ( 10%). Diagnóstico : La mayor sensibilidad la tienen las metanefrinas plasmáticas libres (S 99%), seguidas de las metanefrinas urinarias fraccionadas (S 97%) (orina de 24hs). La determinación menos sensible fue la de AVM (S 64%) pero a su vez las mas especifica (E 95%).")
50
Feocromocitoma RMN – TAC c/ Contraste – Gamagrafía con MIBG: útiles para localización anatómica. Tratamiento: Si es Unilateral de elección es la Adrenalectomía completa. Si es Bilateral debe someterse a una suprarrenalectomía con respeto cortical y a una suprarrenalectomía contralateral total. Conservar la corteza en un lado en lugar de en los dos tiene un riesgo bajo de recidiva del feocromocitoma, y posibilita la independencia de los corticoides en muchos pacientes La preparación farmacológica preoperatoria es un factor clave en la reducción de la morbi-mortalidad. Pronostico : 16% Reicidiva. El seguimiento clínico y bioquímico debe ser indefinido. En la enfermedad maligna la tasa de supervivencia a los 5 años es menor del 50%

51
Bibliografia Bravo EL.pheochromocytoma: new concepts and future trends. Kidney int 1991; 40:544-56 Callender G, Rich T, Perrier N, MD. Sindromes de neoplasia endocrina multiple. Surg Clin N Am 88 (2008) 863–895 Kudva YC, Sawka AM, Young WF Jr. the laboratory diagnosis of adrenal pheochromocytoma: the Mayo Clinic wxperience. J Clin Endocrinol Metab 2003; 88: Pacak K, Eisenhofer G, Alman H, Bornstein SR, et al. Pheochromocytoma: recommendations for clinical practice from the First International Symposium. October Nat Clin Pract Endocrinol Metab 2007; 3: Rodriguez JM, Balsalobre M, Ponce JL, Rios A, et al. pheochromocytoma in MEN 2A syndrome. Study of 54 patientes. World J surg 2008; 32:2520-6 Schartz GL. Screening for adrenal-endocrine hypertension: overview of accurancy and cost-effectiveness. Endocrinol Metab Clin North Am 2011 Jun: 40 (2):
 863–895. Kudva YC, Sawka AM, Young WF Jr. the laboratory diagnosis of adrenal pheochromocytoma: the Mayo Clinic wxperience. J Clin Endocrinol Metab 2003; 88: Pacak K, Eisenhofer G, Alman H, Bornstein SR, et al. Pheochromocytoma: recommendations for clinical practice from the First International Symposium. October Nat Clin Pract Endocrinol Metab 2007; 3: Rodriguez JM, Balsalobre M, Ponce JL, Rios A, et al. pheochromocytoma in MEN 2A syndrome. Study of 54 patientes. World J surg 2008; 32: Schartz GL. Screening for adrenal-endocrine hypertension: overview of accurancy and cost-effectiveness. Endocrinol Metab Clin North Am 2011 Jun: 40 (2):")
Presentaciones similares