Descargar la presentación
La descarga está en progreso. Por favor, espere

1
Reunión Clínica: Distrofia Miotónica
Dr. Carlos Valdebenito Parra Becado Pediatría APS Dr. Guillermo Guzmán Neurólogo Infantil Viernes 20 de diciembre 2013

2
Cronograma Introducción Historia Clasificación Epidemiología Genética
Clínica : Distrofia miotónica congénita, infantil, forma adulta Diagnóstico Manejo Controles

3
Introducción La distrofia muscular más frecuente
Debilidad, miotonía y cataratas son los 3 síntomas cardinales de DM1 Introducción La distrofia muscular más frecuente Enfermedad de intrigante biología molecular , motivo de estudio permanente Cualquier edad Enfermedad multisistémica Deterioro músculo liso, sistemas nervioso y endocrino, ocular, hueso, piel, aparato respiratorio y los sistemas inmunitario y hematopoyético

4
Historia Miopatía miotónica proximal (PROMM) 1995 Ricker
Reconocida por Steinert , Batten y Gibb en 1902, como entidad distinta a Miotonía congénita Deterioro muscular progresivo, miotonía, desórdenes extramusculares Atrofia testicular (Steinert 1909) Cataratas ( Greenfield 1911) Descripción de expresión fenotípica variable : Fleischer Múltiples investigaciones 2001: fenómeno de anticipación Actualmente en investigación terapia génica. Miopatía miotónica proximal (PROMM) 1995 Ricker 1998 se denomina Distrofia miotónica tipo 2 Romeo Vincenzo, MYOTONIC DYSTROPHY TYPE 1 OR STEINERT’S DISEASE, Neurodegenerative Diseases ©2012
 Cataratas ( Greenfield 1911) Descripción de expresión fenotípica variable : 1918 Fleischer. Múltiples investigaciones. 2001: fenómeno de anticipación. Actualmente en investigación terapia génica. Miopatía miotónica proximal (PROMM) 1995 Ricker se denomina Distrofia miotónica tipo 2. Romeo Vincenzo, MYOTONIC DYSTROPHY TYPE 1 OR STEINERT’S DISEASE, Neurodegenerative Diseases ©2012.")
5
Distrofia Distrofia muscular: Miotonía
Del griego dis que significa difícil o defectuoso y trof nutrición Distrofia muscular: Grupo de enfermedades genéticas que causan debilidad y degeneración progresiva de músculos esqueléticos. Miotonía Desorden caracterizado por dificultad y retraso en relajación del músculo esquelético ,después de la contracción voluntaria o efecto de percusión

7
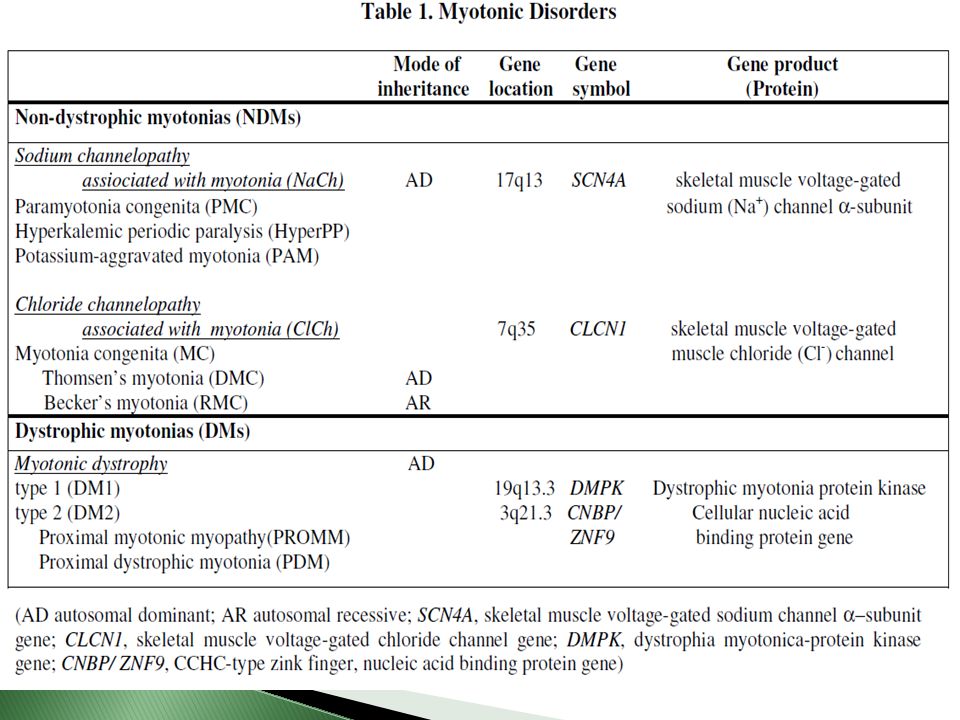
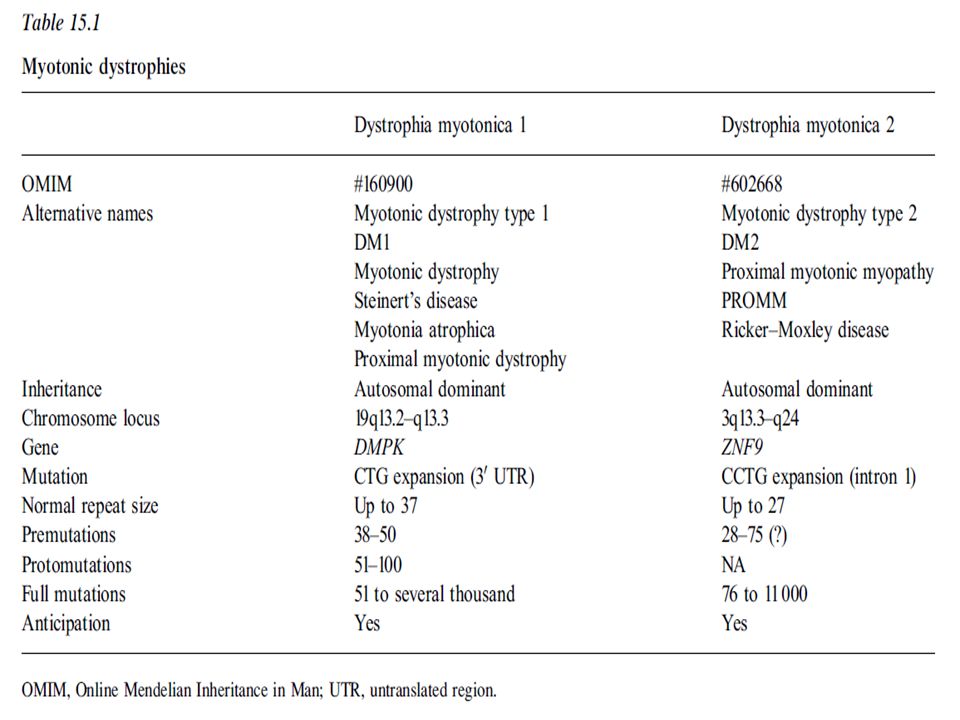
Clasificación Distrofia miotónica Tipo 1 Infantil Tipo 2 Adulta
Congénita Infantil Tipo 2 Adulta Oligosintomática The myotonic dystrophies: molecular, clinical, and therapeutic challenges Bjarne Udd, Ralf Krahe Lancet Neurol 2012; 11: 891–905

8
Epidemiología Previo a identificación de ambos tipos se estimaba en 1/8000 (Prevalencia 12,5/ ) a nivel mundial. Varía según la población , más alta en Canadá, Región Basca de España, Finlandia. TIPO 1 1/ habitantes nivel mundial 1/8000 Europa Regiones Canadá (Quebec) prevalencia 189/ Pacientes jóvenes con cataratas, 0.9-3% tienen mutación DM1 TIPO 2 1/ Finlandia: Mutación: 1/ (tipo 1 prevalencia 1/2760) The myotonic dystrophies: molecular, clinical, and therapeutic challenges Bjarne Udd, Ralf Krahe Lancet Neurol 2012; 11: 891–905
 prevalencia 189/ Pacientes jóvenes con cataratas, 0.9-3% tienen mutación DM1. TIPO 2. 1/ Finlandia: Mutación: 1/1830 (tipo 1 prevalencia 1/2760) The myotonic dystrophies: molecular, clinical, and therapeutic challenges. Bjarne Udd, Ralf Krahe Lancet Neurol 2012; 11: 891–905.")
9
RN con hipotonía severa: 12-16% DMC
Series pequeñas : 1/ (no estudios poblacionales) Otros estudios 2,1/ Incidencia Cerca de 48% Rn diagnosticados corresponden a caso índice familiar Neurology and Neurophysiology 2012, Campbell, Congenital Myotonic Distrophy
 Otros estudios ,1/ Incidencia. Cerca de 48% Rn diagnosticados corresponden a caso índice familiar. Neurology and Neurophysiology 2012, Campbell, Congenital Myotonic Distrophy.")
10
Genética Autosómica dominante
Penetrancia casi completa y expresividad variable Fenómeno de anticipación Influencia del sexo del padre afectado : Distrofia congénita se asocia a madre afectada Neurology and Neurophysiology 2012, Campbell, Congenital Myotonic Distrophy

11
Fenómeno de anticipación Distrofia Miotónica

12
Todas las expansiones llevan a inestabilidad
Distrofia Miotónica Tipo 1 98% Amplificación de triplete CTG en el extremo 3´ no transcrito de un gen situado en cromosoma 19 q13.3, codificante para proteína DMPK de las familias de proteincinasas. (Distrophy Miotonic protein kinasa) Kda Función: Forma celular y homeostasis del calcio (excitación celular) Inestabilidad somática : el tamaño de expansión no es igual en todos los tejidos (mayor en gametos) Todas las expansiones llevan a inestabilidad Neurology and Neurophysiology 2012, Campbell, Congenital Myotonic Distrophy
 Kda. Función: Forma celular y homeostasis del calcio (excitación celular) Inestabilidad somática : el tamaño de expansión no es igual en todos los tejidos (mayor en gametos) Todas las expansiones llevan a inestabilidad. Neurology and Neurophysiology 2012, Campbell, Congenital Myotonic Distrophy.")
13
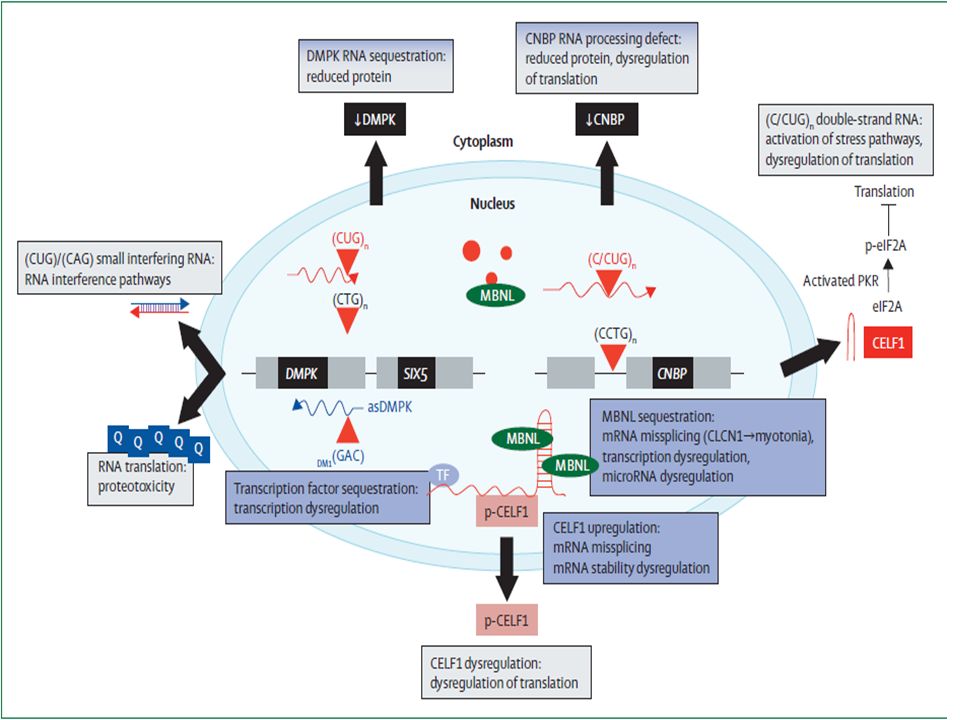
DMPK mutante Dos dominios catalíticos adicionales Genera en su transcripción toxicidad celular Estas isoformas Peso entre 80 kD y 86 kD Se agrupa en la membrana de la unión neuromuscular y del retículo sarcoplásmico Relación alterada con miosina Ausencia de DMPK favorece la forma hipofosforilada de CUG-BP que se acumula en el núcleo. Gen mutado se transcribe pero ARNm se retiene en el núcleo. Déficit de proteína Acumulación de este mRNA tiene efectos citotóxicos. ARN m DMPK se expresa en músculo esquelético, corazón, cerebro, hígado y riñones Inhibe normalmente Miosina fosfatasa , y regula tamaño y forma celular

14
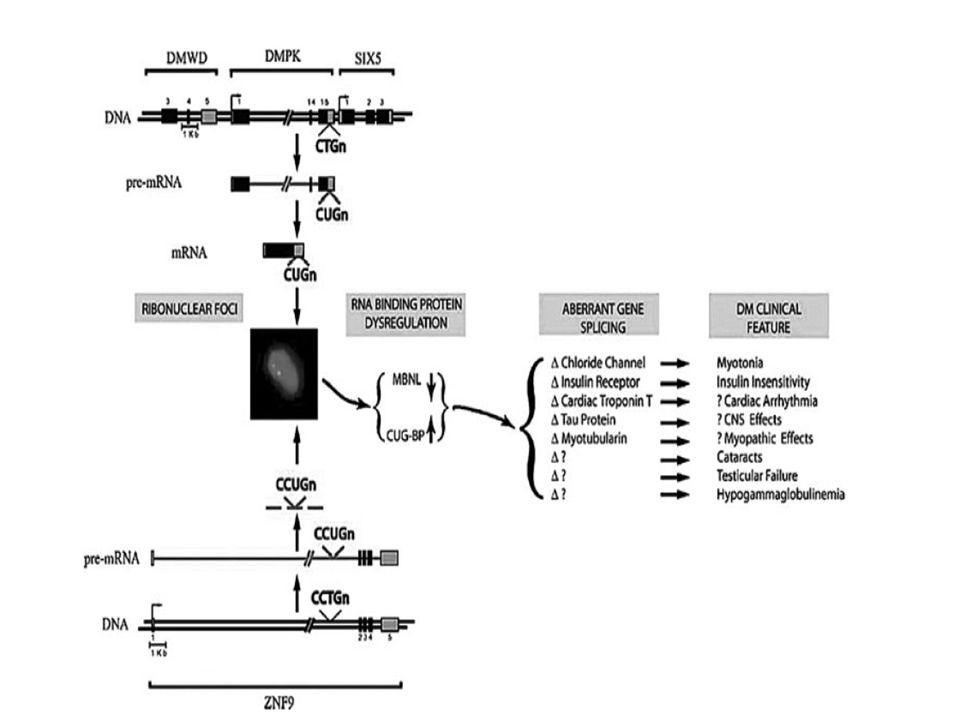
Factores genéticos Condensación anómala de la cromatina
Modificación de la cromatina en la región donde se encuentra el trinucleótido repetido altera la expresión de genes adyacentes. Genes próximos al locus DMPK, DMWD (río arriba) y SIX5 (río abajo) se transcriben menos en presencia de expansiones muy largas de CTG Alteración de proteínas de unión a ARNm lleva a la formación de focos tóxicos de mRNA y a un splicing aberrante. Medwave 2005 Abr;5(3):e3370 doi: /medwave Myotonic dystrophy: genetic aspects
 y SIX5 (río abajo) se transcriben menos en presencia de expansiones muy largas de CTG. Alteración de proteínas de unión a ARNm lleva a la formación de focos tóxicos de mRNA y a un splicing aberrante. Medwave 2005 Abr;5(3):e3370 doi: /medwave Myotonic dystrophy: genetic aspects.")
15
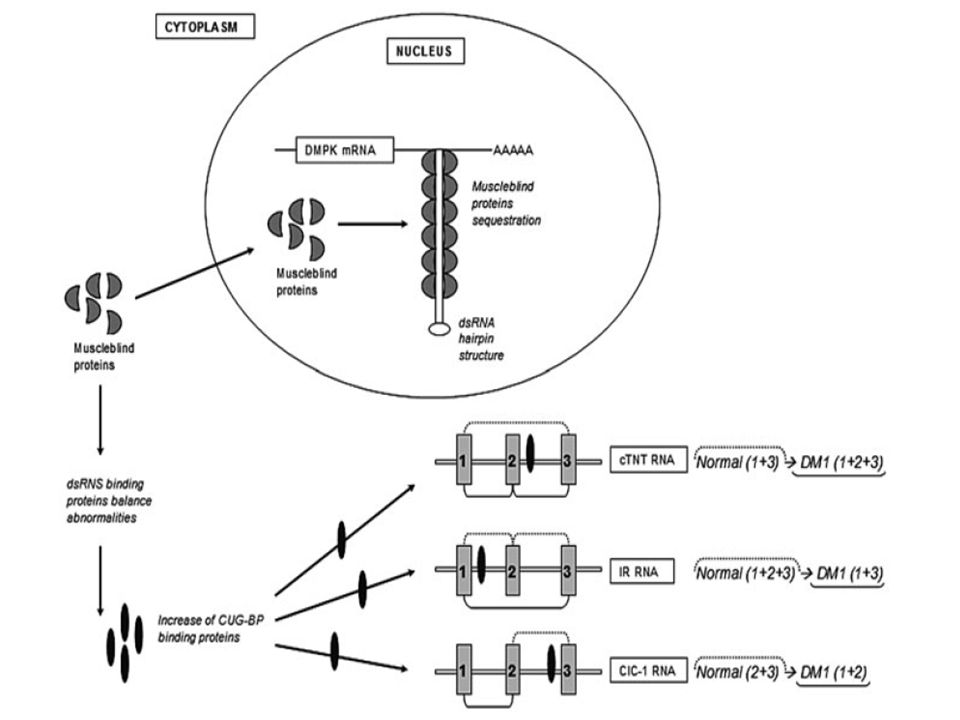
MBNL (muscle blind proteins ) 1, 2 y 3 son secuestradas por ARN mutante (CUG-ARN)
CUG –BP (Binding proteins) tienden a tener mayor actividad ALTERACION DE DISTINTOS RECEPTORES Y CANALES que explican manifestaciones clínicas NMDA en cerebro, subunidad NR1, proteína tau, proteína precursora de amiloide (APP). Canal de cloro Clcnl, miotonía
 tienden a tener mayor actividad. ALTERACION DE DISTINTOS RECEPTORES Y CANALES que explican manifestaciones clínicas. NMDA en cerebro, subunidad NR1, proteína tau, proteína precursora de amiloide (APP). Canal de cloro Clcnl, miotonía.")
17
ARNm alterados en DM codifican proteínas alteradas
Proteína Tau Proteína relacionada con la miotubularina 1 (MTMR1): fenotipo, atrofia, debilidad ARNm codificante para el receptor de insulina (IR) (isoforma A predominante en desmedro de B) ARNm codificante para troponina T cardíaca (cTNT) : arritmogénica ARNm codificante para el canal de cloro 1 (ClC-1).
: fenotipo, atrofia, debilidad. ARNm codificante para el receptor de insulina (IR) (isoforma A predominante en desmedro de B) ARNm codificante para troponina T cardíaca (cTNT) : arritmogénica. ARNm codificante para el canal de cloro 1 (ClC-1).")
19
Pérdida de función de SIX5 Pérdida de función de DMWD
Involucrada en desarrollo muscular y gonadal Asociación con cataratas Regula función de subunidad a1 de NaKATPasa. Desbalance osmótico, opacidad. Teoría de alteración cardíaca Pérdida de función de DMWD Cerebro y gónadas Asociación con daño muscular .

20
Distrofia Miotónica Tipo 2
2% PROMM (“proximal myotonic myopathia”) Su causa es la expansión de tetranucléotido CCTG en gen 3q13.3- q24 Proteína CNBP ,nucleic acid binding protein gene (previamente llamado ZNF9) Normal: 10-30 Afectados: Escasa anticipación, sin formas congénitas severas, sin origen parental específico, sin correlación con la edad. Chen Sun, Clinical and genetic investigations of patients with myotonia congenita in Northern Norway, 2011, FACULTY OF HEALTH SCIENCES DEPARTMENT OF CLINICAL MEDICINE - MEDICAL GENETICS
 Su causa es la expansión de tetranucléotido CCTG en gen 3q13.3- q24. Proteína CNBP ,nucleic acid binding protein gene (previamente llamado ZNF9) Normal: Afectados: Escasa anticipación, sin formas congénitas severas, sin origen parental específico, sin correlación con la edad. Chen Sun, Clinical and genetic investigations of patients. with myotonia congenita in Northern Norway, 2011, FACULTY OF HEALTH SCIENCES DEPARTMENT OF CLINICAL MEDICINE - MEDICAL GENETICS.")
24
Clínica

25
Distrofia miotónica congénita
Descrita en 1960. Transmisión por vía materna de manera casi exclusiva Prenatal: Aborto Polihidroamnios Disminución de movimientos fetales Diafragma hipoplásico Costillas delgadas Hidrocefalia Artrogriposis

26
Distrofia miotónica congénita: post natal
Sistema sensorial normal Puede asociar encefalopatía hipóxico isquémica, que puede enmascarar el diagnóstico Alteración succión deglución , lenta evolución de la alimentación, uso de SNG, requerimiento de procinéticos, antiácidos. Si sobreviven primeros meses: mejoría progresiva, con retraso mental, variable aparición de manifestaciones de forma clásica Letalidad 16% Hipotonía e hipoactividad generalizada/ Bajos puntajes de APGAR Debilidad proximal y distal Diplejía facial Alteraciones respiratorias , Hipertensión pulmonar persistente

27
Mortalidad 16-41% (respiratorias, arritmias)
80% niños tendrán arritmia, taquicardia, PR prolongado, defectos conducción intranodal Estrabismo ¼ Cataratas son de aparición más tardía

28
Diagnóstico diferencial
Neonatos Atrofia muscular espinal Distrofias musculares congénitas Miopatías congénitas Diversas encefalopatías Disha, Pediatric Neurology 2012,The Spectrum of Myotonic and Myopathic Disorders in a Pediatric Electromyography Laboratory Over 12 Years

29
Distrofia miotónica infantil
Sin Miotonía . Generalmente aparece post 8-10 años. Normales al nacimiento Fenotipo intermedio (congénito-adulto) Debilidad facial : boca en tienda Se acrecienta a medida que avanza la edad Problemas de lenguaje y de alimentación Disartria Dificultades en cuidado dental Primera década de la vida, estables con retraso de hitos del desarrollo pero escasa mortalidad EMG: descargas miotónicas Segunda década de la vida Miotonía se hace evidente Debilidad muscular, contracturas musculares, cifoescoliosis (10-30%) Retraso mental variable Comorbilidad con SDAH, déficits función ejecutiva , trastornos de ansiedad y autismo (hasta 50%)
 Debilidad facial : boca en tienda. Se acrecienta a medida que avanza la edad. Problemas de lenguaje y de alimentación. Disartria. Dificultades en cuidado dental. Primera década de la vida, estables con retraso de hitos del desarrollo pero escasa mortalidad. EMG: descargas miotónicas. Segunda década de la vida. Miotonía se hace evidente. Debilidad muscular, contracturas musculares, cifoescoliosis (10-30%) Retraso mental variable. Comorbilidad con SDAH, déficits función ejecutiva , trastornos de ansiedad y autismo (hasta 50%)")
30
Miotonía DM1 DM2 No dolorosa, molesta
Exacerbada por frío (warm up phenomenon) De acción y percusión Indetectable en niños pequeños Disminuye a medida que aumenta atrofia Alteraciones canales Cloro, Sodio y K. DM1 DM2
 De acción y percusión. Indetectable en niños pequeños. Disminuye a medida que aumenta atrofia. Alteraciones canales Cloro, Sodio y K. DM1. DM2.")
31
Sin relación con tipo de transmisión ni con extensión de triplete
Objetivo: Uso de escalas diagnósticas en población de niños con DM1 para evaluar cognición, patología psiquiátrica y correlación con tamaño de la expansión de triplete en Francia 28 niños entraron al estudio (dx confirmado, historia prenatal y postnatal sin eventos , desarrollo normal primer año) Grupo menores de 18 años (11) 18-24 años (17) Resultados: 19/28 repitencia escolar, 11 educación diferencial, 17 adultos mal desempeño laboral. Ci armónico 15 pacientes con diagnósticos DSM IV: Fobia (7/ 25%), desorden del ánimo (6/21%), ansiedad (5/ 18%), ) tx oposicionista desafiante (5/18%). SDAH (8/ 29%), sin autismo ni psicosis Sin relación con tipo de transmisión ni con extensión de triplete
 Grupo menores de 18 años (11) años (17) Resultados: 19/28 repitencia escolar, 11 educación diferencial, 17 adultos mal desempeño laboral. Ci armónico. 15 pacientes con diagnósticos DSM IV: Fobia (7/ 25%), desorden del ánimo (6/21%), ansiedad (5/ 18%), ) tx oposicionista desafiante (5/18%). SDAH (8/ 29%), sin autismo ni psicosis. Sin relación con tipo de transmisión ni con extensión de triplete.")
32
Importancia de síntomas psiquiátricos y cognitivos en niños y adolescentes con DM1.
Importancia de la sospecha , y del manejo multidisciplinario

33
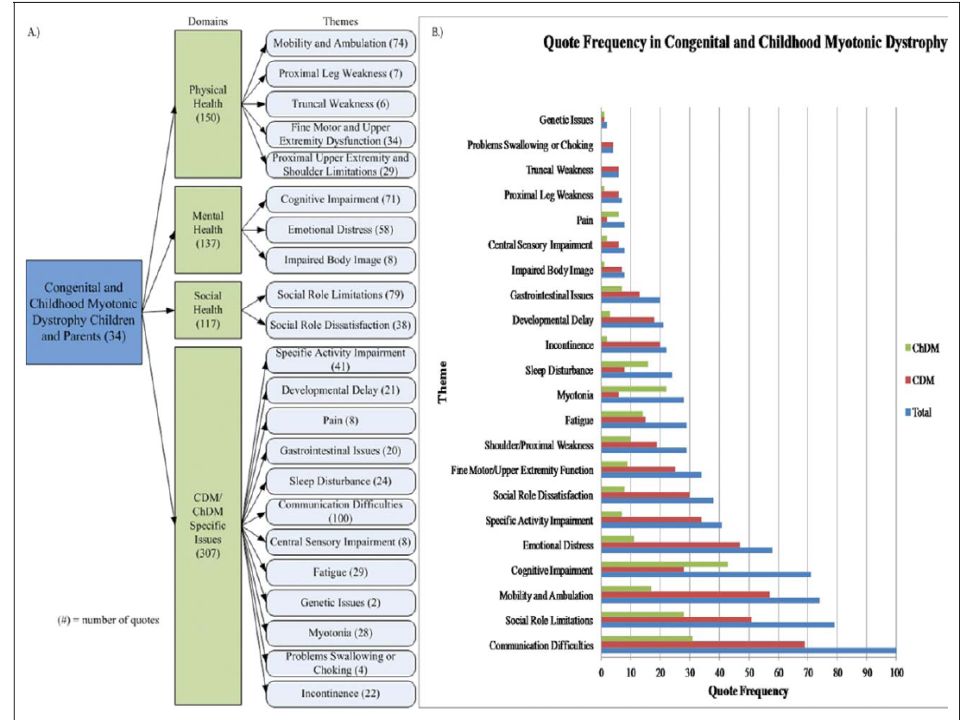
Entrevistas a padres y pacientes con distrofia miotónica congénita e infantil.
Universidad de Rochester Desarrollar modelo representativo de salud y calidad de vida Metodología: niños >8 años , evaluación síntomas físicos, mentales y sociales Resultados: Entrevista a 34 familias, 189 síntomas identificados. Dificultades comunicación e interacción social Dificultades aprendizaje, problemas de concentración, síntomas emocionales

35
Clínica : forma adulta Manifestaciones musculares Debilidad muscular, miotonía. Debilidad de músculos faciales, temporales, compromiso de músculos orbiculares, blefaroptosis bilateral. “Hatchet face” Fascie miopática: inexpresividad Rigidez facial, hipomimia, cabello escaso. Disfagia orofaríngea, riesgo de aspiración Disartria Cuello, debilidad distal de extremidades Debilidad, miotonía y cataratas son los 3 síntomas cardinales de DM1

36
Manifestaciones musculares faciales

38
Compromiso cardíaco Asintomático hasta muerte súbita
Subestimado en niños Patología cardíaca aparece precozmente en DM 1 tipo congénita o de la niñez Adultos asintomáticos: 29% disfunción sistólica Fibrosis miocárdica y degeneración del sistema excitoconductor Cardiomiopatía Taquiarritmias: FA, fibrilación auricular (30%) Taquicardias ventriculares Fibrosis en sistema de conducción
 Taquicardias ventriculares. Fibrosis en sistema de conducción.")
39
11% fallecimiento (solo 1 por MS)
5,7 años de seguimiento 20 % requirió marcapasos 2.7% DAI 11% fallecimiento (solo 1 por MS) Facenda-Lorenzo M, et al. Manifestaciones cardiacas en los pacientes con distrofia miotónica tipo 1 seguidos de forma protocolizada en una consulta monográfica. Rev Esp Cardiol. 2012
 Facenda-Lorenzo M, et al. Manifestaciones cardiacas en los pacientes con distrofia miotónica tipo 1 seguidos de forma protocolizada en una consulta monográfica. Rev Esp Cardiol")
40
Compromiso respiratorio
1) Aspiración 2) Atrofia muscular Debilidad diafragmática e intercostal Deformidad esquelética Hipoventilación 3)Alteración coordinación respiratoria SNC Requiere precauciones anestésicas, 40% pacientes adultos riesgo de complicaciones Petrica, Functional and histopathological identification of the respiratory failure in a DMSXL transgenic mouse model of myotonic dystrophy, 2013, Disease models and mechanisms
 Aspiración. 2) Atrofia muscular. Debilidad diafragmática e intercostal. Deformidad esquelética. Hipoventilación. 3)Alteración coordinación respiratoria SNC. Requiere precauciones anestésicas, 40% pacientes adultos riesgo de complicaciones. Petrica, Functional and histopathological identification of the respiratory failure in a DMSXL transgenic mouse model. of myotonic dystrophy, 2013, Disease models and mechanisms.")
41
Somnolencia diurna: 70-80% adultos con DM1. Hasta 50% en pediatría.
Multifactorial, baja secreción pulsátil de cortisol y hormona de crecimiento. Aumento de IL-6 y TNF alfa : proinflamación y somnífero Gliosis formación reticular Fragmentación del sueño Comorbilidad con depresión es mayor Peor evolución muscular que pacientes sin trastorno y peor calidad de vida Hasta 22% adultos síndrome de piernas inquietas. Trastornos respiratorios del sueño: compromiso músculo respiratorios, anormalidades del control central. (25% pacientes al inicio -77% etapas finales tienen hipercapnia) Indice apneas/hipopneas alterado
 Indice apneas/hipopneas alterado.")
42
Anormalidades SNC Estructurales y funcionales
Alteraciones sustancia blanca y en sustancia gris Acumulación de ovillos neurofibilares (proteína tau) y cuerpos de inclusión En adultos subestimada , disfunción ejecutiva, apatía, personalidad evitativa, alteraciones de la calidad de vida Desórdenes psiquiátricos Alteraciones del sueño Alteraciones sensoriales : adultos hasta 62,5% compromiso auditivo leve-severo (Balatsouras Inner ear dysfunction in myotonic dystrophy type 1, 2012, acta neurologica Scandaniva)
 y cuerpos de inclusión. En adultos subestimada , disfunción ejecutiva, apatía, personalidad evitativa, alteraciones de la calidad de vida. Desórdenes psiquiátricos. Alteraciones del sueño. Alteraciones sensoriales : adultos hasta 62,5% compromiso auditivo leve-severo. (Balatsouras Inner ear dysfunction in myotonic dystrophy type 1, 2012, acta neurologica Scandaniva)")
43
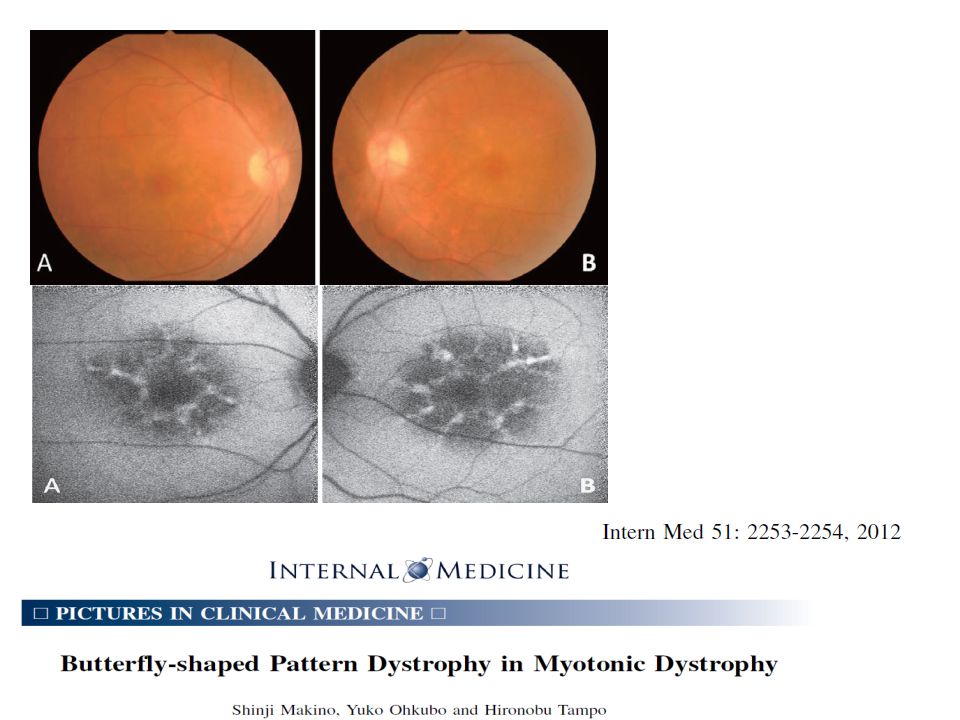
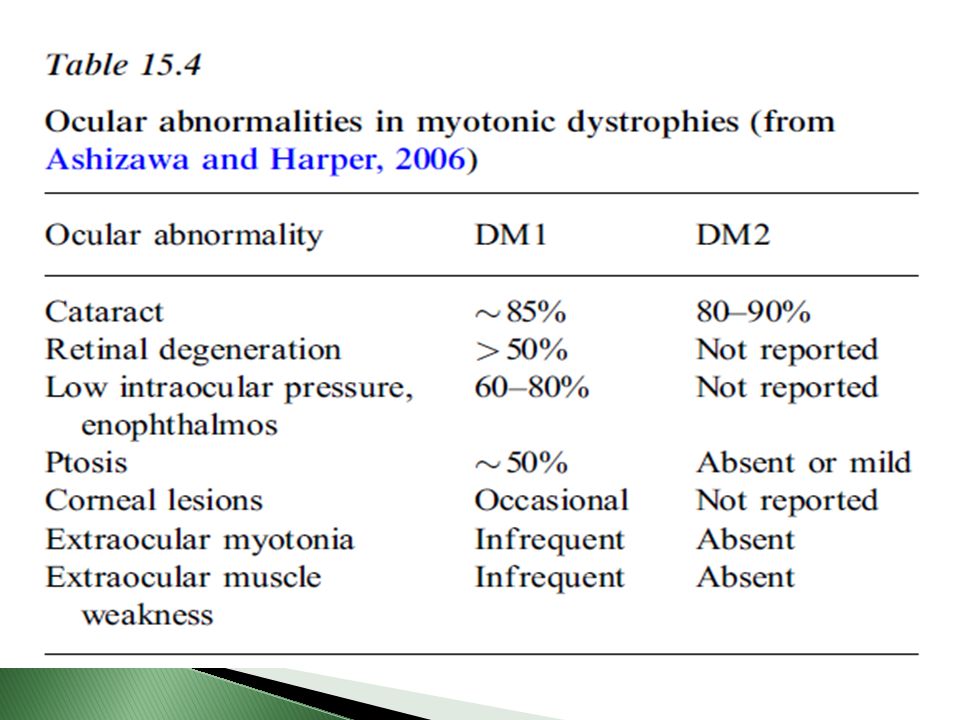
Alteraciones oculares
Cataratas Se incrementa a medida que avanza la edad (80% llega a tenerlas) Etapas tempranas, apariencia multicolor en región subcapsular De alerta para dx menores de 50 años Alteraciones de retina: mácula y periféricas Ptosis Debilidad músculos orbiculares, hipotensión ocular Miotonía en músculos oculares, más presente en congénita Hipermetropía, ambliopía.
 Etapas tempranas, apariencia multicolor en región subcapsular. De alerta para dx menores de 50 años. Alteraciones de retina: mácula y periféricas. Ptosis. Debilidad músculos orbiculares, hipotensión ocular. Miotonía en músculos oculares, más presente en congénita. Hipermetropía, ambliopía.")
46
Alteraciones gastrointestinales
Disfagia por alteración motilidad y por disminución presión esfínter esofágico superior, RGE Alteraciones deglución (hasta 30% adultos) Regurgitación, dispepsia, dolor abdominal, cambios hábito intestinal, diarrea 30-40%. Vaciamiento gástrico enlentecido 28%. Megacolon, pseudoobstrucción intestinal en niños y adultos. Complicaciones : Ileo, vólvulo, perforación colónica . Malabsorción. Translocación bacteriana Hay reportes de niños con Colelitiasis . Alteración vaciamiento biliar Umemoto, Dysphagia in DMD vs. DM1 MUSCLE & NERVE October 2012
 Regurgitación, dispepsia, dolor abdominal, cambios hábito intestinal, diarrea 30-40%. Vaciamiento gástrico enlentecido 28%. Megacolon, pseudoobstrucción intestinal en niños y adultos. Complicaciones : Ileo, vólvulo, perforación colónica . Malabsorción. Translocación bacteriana. Hay reportes de niños con Colelitiasis . Alteración vaciamiento biliar. Umemoto, Dysphagia in DMD vs. DM1 MUSCLE & NERVE October")
48
Compromiso hepático Sin fisiopatología establecida

49
Anomalías endocrinas

50
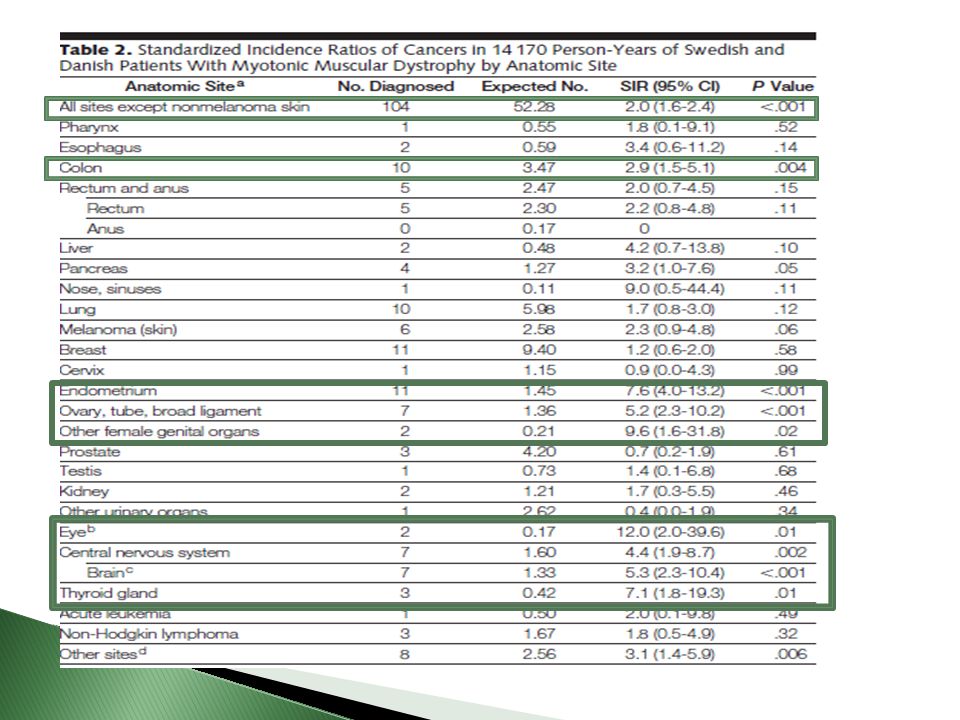
Cáncer Estudio: 1658 pacientes con DM Hospital Suizo 1977-2008
104 pacientes (73.4/ ), doble de lo esperado poblacionalmente Endometrio, Ovario, cerebro y colon Epiteliomas y pilomatrixomas Shahinaz, Cancer Risk Among Patients With Myotonic Muscular Dystrophy JAMA, December 14, 2011—Vol 306,
, doble de lo esperado poblacionalmente. Endometrio, Ovario, cerebro y colon. Epiteliomas y pilomatrixomas. Shahinaz, Cancer Risk Among Patients. With Myotonic Muscular Dystrophy JAMA, December 14, 2011—Vol 306,")
52
Estudio retrospectivo 21 pacientes con DM1 Alemania
17 pacientes (81%) Dm tipo 1 tipo congénita 4 pacientes (19%) forma adulta Promedio edad 15,3 años Contracturas: 52% (10% extremidades superiores, 48% extremidades inferiores) Deformidades pies: 20 pacientes (95%), equino más frecuente, cavo 2°lugar Fracturas: 5 pacientes (24%)
 Dm tipo 1 tipo congénita. 4 pacientes (19%) forma adulta. Promedio edad 15,3 años. Contracturas: 52% (10% extremidades superiores, 48% extremidades inferiores) Deformidades pies: 20 pacientes (95%), equino más frecuente, cavo 2°lugar. Fracturas: 5 pacientes (24%)")
53
43% deformidades espinales

54
Distrofia miotónica tipo 2
Más tenue y menos progresiva que tipo 1 Debilidad y atrofia proximal , debilidad de músculos flexores profundos de los dedos Menos compromiso facial y bulbar Dolor, hipertrofia muscular , mialgias, fatiga Miotonía más difícil de encontrar Menor frecuencia de arritmias severas Menos frecuencia de anticipación

55
Diagnóstico Clínica: Dependiendo de la edad Exámenes generales
CK: puede estar normal o elevación máxima 1000 U/L Aumento de GGT Hipogamaglobulinemia G Alteraciones hormonales en adultos

56
Diagnóstico Electromiografía Miotonía eléctrica Patrón miopático
VCN normal

57
Histología forma congénita:
Sin evidencia de distrofia Núcleos centrales y halos sarcoplásmicos periféricos en pequeñas fibras Reducción de elementos contráctiles en músculo esquelético y cardíaco Alteración en Myotubularin Related Protein 1 (MTMR1). Relacionado en maduración muscular. Reducida densidad de canales de sodio.
. Relacionado en maduración muscular. Reducida densidad de canales de sodio.")
58
Biopsia Típica pero no patognomónica
Variación en tamaño de las fibras musculares con atrofia de fibras tipo I. Núcleo central, fibras en anillo, masas sarcoplásmicas, fibras necróticas Aumento de tejido conectivo, rara vez hay reemplazo graso DM1 DM 2

59
Diagnóstico Genético Prenatal Southern Blot
Evaluación de expansión. Falsos (-) por baja de sensibilidad por heterogeneidad somática Reacción cadena de polimerasa (<100 repeticiones) Hibridización in situ con fluorescencia (FISH) Hibridización in situ cromogénica (CISH) Prenatal Tamaño del alelo expandido, > de riesgo para DM congénita a través de muestra de amniocentesis o vellosidad coriónica
 por baja de sensibilidad por heterogeneidad somática. Reacción cadena de polimerasa (<100 repeticiones) Hibridización in situ con fluorescencia (FISH) Hibridización in situ cromogénica (CISH) Prenatal. Tamaño del alelo expandido, > 1000 de riesgo para DM congénita a través de muestra de amniocentesis o vellosidad coriónica.")
60
Imágenes RNM muscular Cambios degenerativos van paralelos a la clínica de debilidad y atrofia DM1 : compromiso distal sóleo y gastrocnemio DM2: grupo vasto anterior , recto femoral

61
RNM cerebral Morfología, perfusión (SPECT) , metabólico (PET)
Congénita: Grados variables de atrofia cerebral, ventriculomegalia e hiperintensidad T2 Similar a infección por CMV (multifocal con calcificaciones periventriculaes y lesiones quísticas) Defectos del desarrollo Formas adultas: Dilatación ventricular no progresiva Moderada o grave afectación de la sustancia blanca posterosuperior a los trígonos Compromiso variable de sustancia blanca, cápsulas interna y externa , prefrontal. Morfología, perfusión (SPECT) , metabólico (PET) Ravikanth, 2012, Magnetic resonance imaging findings in adult form in myotonic dystrophy 1, Singapore Med J
 Defectos del desarrollo. Formas adultas: Dilatación ventricular no progresiva. Moderada o grave afectación de la sustancia blanca posterosuperior a los trígonos. Compromiso variable de sustancia blanca, cápsulas interna y externa , prefrontal. Morfología, perfusión (SPECT) , metabólico (PET) Ravikanth, 2012, Magnetic resonance imaging findings in adult form in myotonic dystrophy 1, Singapore Med J.")
62
Manejo Sin terapia específica aún. Manejo de síntomas y complicaciones
Problemas musculares Miotonía Eventual uso de fenitoína, carbamazepina, procainamida mexiletina (modula canales de sodio) Terapia física Terapia ocupacional Manejo Sin terapia específica aún. Manejo de síntomas y complicaciones Dolor: AINES, antidepresivos tricíclicos, bajas dosis de corticoides Debilidad DHEA: sin evidencia para su uso IGF-1: mejora sensibilidad insulina, anabolismo muscular, pero requeria inyecciones 2 veces semana. No uso actual. Creatina : distrofias, pequeño beneficio en 8.8% mejora de contracción voluntaria máxima. Aumenta ATP intracelular.
 Terapia física. Terapia ocupacional. Manejo. Sin terapia específica aún. Manejo de síntomas y complicaciones. Dolor: AINES, antidepresivos tricíclicos, bajas dosis de corticoides. Debilidad. DHEA: sin evidencia para su uso. IGF-1: mejora sensibilidad insulina, anabolismo muscular, pero requeria inyecciones 2 veces semana. No uso actual. Creatina : distrofias, pequeño beneficio en 8.8% mejora de contracción voluntaria máxima. Aumenta ATP intracelular.")
63
Manejo interdisciplinario
Manejo Respiratorio Manejo Cardiológico Según condición clínica Arritmias: Uso de b- bloqueadores , amiodarona Implante de desfibrilador/marcapasos Período neonatal Manejo ventilatorio, reanimación Uso de Cpap/ VM Aminofilina/ Cafeína para manejo de apneas Higiene del sueño, manejo ambiental Evaluación de hipoventilación: debilidad diafragmáticas BIPAP o CPAP nocturno Prevención de complicaciones perioperatorias Fármacos contraindicados: tiopental, neostigmina, halotano. Precaución narcóticos y benzodiacepinas.

64
Manejo interdisciplinario
Problemas de SNC Programas educacionales para niños Metilfenidato Modafinilo Antidepresivos/ Ansiolíticos Problemas oculares Remoción de cataratas, blefaroplastia. Problemas digestivos Alimentación saludable Alto contenido de fibra Manejo farmacológico de constipación Procinético? Eritromicina? Diarrea: colestiramina Manejo eventual de patología vesicular Pennock, Therapeutics Development in Myotonic Dystrophy Type I, Muscle nerve 2012

65
Terapia específica Degradación de ARNm tóxico con expansión Ribozimas 2003: Puymirat publicó estudio con reducción de 63% de forma mutante, pero con reducción también de ARN normal en 50% con ribozimas Oligonucleótido antisensor 2′-O-methyl modified AONs (MOE)/ Phosphorodiamidate morpholino antisense molecules (PMO) Sólo se ha probado con efecto local (No sistemico) con reducción de sólo 50% Neutralización sitios de unión de proteínas de unión a ARN Pentamidina CAG25 Reducir tamaño de expansión trinucleotídica Bloqueo de acción de MBNL-1y CUG-BP Leung, Therapeutic Advances in Muscular Dystrophy. ANN NEUROL 2013;74:404–411
/ Phosphorodiamidate morpholino antisense molecules (PMO) Sólo se ha probado con efecto local (No sistemico) con reducción de sólo 50% Neutralización sitios de unión de proteínas de unión a ARN. Pentamidina. CAG25. Reducir tamaño de expansión trinucleotídica. Bloqueo de acción de MBNL-1y CUG-BP. Leung, Therapeutic Advances in Muscular Dystrophy. ANN NEUROL 2013;74:404–411.")
66
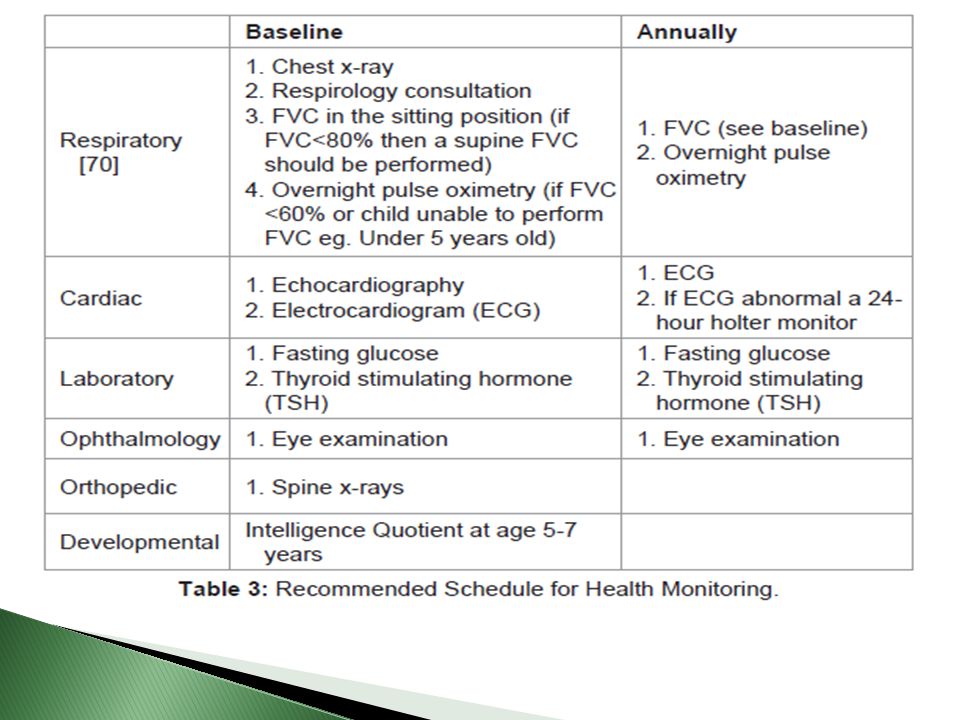
Controles Examen clínico con evaluación neurológica anual
Control cardiológico anual ECG anual Holter de ritmo cada 2 años Ecocardiograma cada 5 años Estudio electrofisiólogico a quienes padezcan de arritmia Examen periódico oftalmológico con fondo de ojo y lámpara de hendidura Control de rehabilitación/ortopedia Control endocrino periódico: evaluación de función tiroidea, función suprarrenal, pubertad tardía, alopecia, atrofia testicular y diabetes Analítica básica con IgG y niveles de colesterol

68
En relación a paciente 18 años
Madre y hermanos afectados de cuadro miopático 10 años. Contracturas , tenotomía aquiliana Debilidad progresiva y generalizada , Escoliosis progresiva , cuadros respiratorios a repetición 14 años: HBIA en ECG/ eco cardio normal 18 años: ex fisico : fascie acorde, contracturas, atrofia muscular, Miotonías + Evolución respiratoria con neumonia grave, Fibrilación auricular,TSV. Cataratas subcapsular Discapacidad intelectual moderada CUADRO COMPATIBLE DISTROFIA MIOTONICA TIPO I

Presentaciones similares










>")











