Descargar la presentación
La descarga está en progreso. Por favor, espere

1
ALTERACIONES EN EL METABOLISMO DE LAS LIPOPROTEINAS
Estructura de las apoproteínas, biosíntesis y metabolismo de lipoproteínas. Clasificación de Fredrickson. Alteraciones en el metabolismo de los Quilomicrones. Deficiencia de lipoproteín lipasa y Síndrome de Quilomicronemia. Estructura, función y deficiencia de LPL y apo CII. Cuadro clínico. Diagnóstico y tratamiento. Deficiencia de Lecitin colesterol acil transferasa. Características de la enzima. Cuadro clínico y anormalidades clínicas y tisulares.

2
ALTERACIONES EN EL METABOLISMO DE LAS LIPOPROTEÍNAS
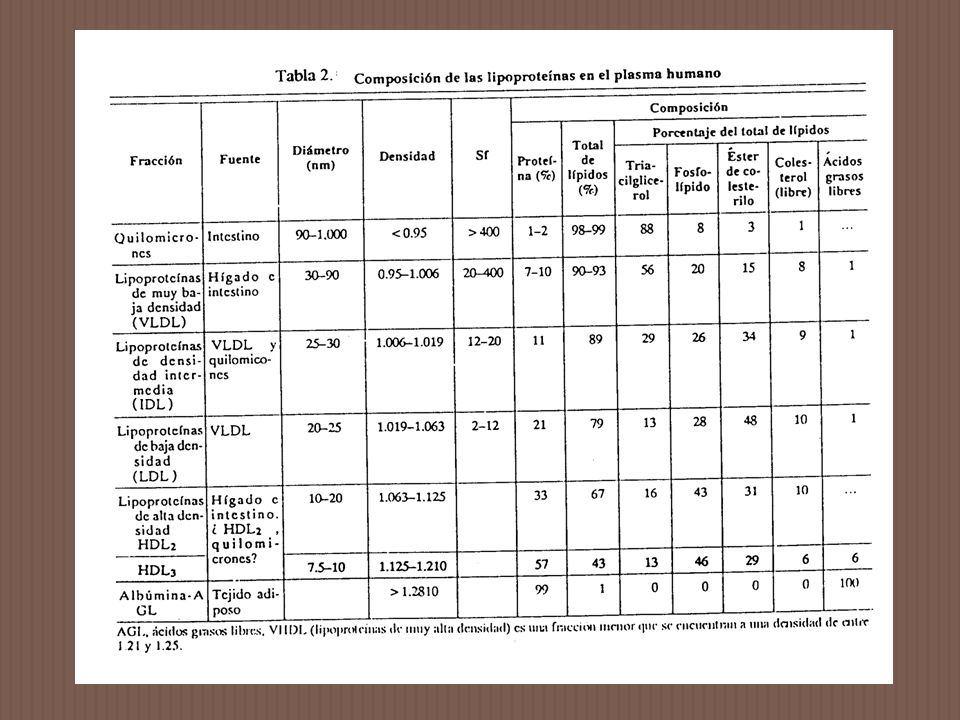
En un omnívoro como el ser humano, se ingieren calorías en exceso en la fase anabólica del ciclo alimentario, seguido por un período de equilibrio calórico negativo en que el organismo utiliza sus reservas de carbohidratos y grasas. Las lipoproteínas (LP) median este ciclo transportando los lípidos del intestino como quilomicrones (Q) y a los hepáticos como lipoproteínas de muy baja densidad (VLDL), para su oxidación en gran parte de los tejidos y al tejido adiposo para su almacenamiento. Los lípidos son movilizados de este último tejido como ácidos grasos libres (AGL) fijados a la albúmina sérica. Las anormalidades del metabolismo de los lípidos se presentan en los sitios de producción o en los de utilización de las LP, causando varios tipos de Hipo e Hiperlipoproteinemias.
 median este ciclo transportando los lípidos del intestino como quilomicrones (Q) y a los hepáticos como lipoproteínas de muy baja densidad (VLDL), para su oxidación en gran parte de los tejidos y al tejido adiposo para su almacenamiento. Los lípidos son movilizados de este último tejido como ácidos grasos libres (AGL) fijados a la albúmina sérica. Las anormalidades del metabolismo de los lípidos se presentan en los sitios de producción o en los de utilización de las LP, causando varios tipos de Hipo e Hiperlipoproteinemias.")
3
La más común es la Diabetes sacarina, donde la deficiencia de Insulina causa la movilización excesiva de AGL y la subutilización de Q y VLDL que conduce a Hipertrigliceridemia. La mayor parte de los demás trastornos patológicos que afectan al transporte lipídico, se deben de manera primaria a Defectos Hereditarios en la síntesis de la porción apoproteica de la LP, de las enzimas clave o de los receptores de LP. Algunos de estos defectos causan Hipercolesterolemia y Aterosclerosis prematura. Los depósitos excesivos de grasa causan la Obesidad, la cual puede deberse a termogénesis defectuosa inducida por la alimentación en el tejido adiposo pardo.

8
FUNCIONES DE LAS PRINCIPALES APOLIPOPROTEINAS
Función A-I Activa la enzima Lecitina Colesterol Acil Transferasa; Rol estructural en la HDL. A-II Papel estructural en algunas subfracciones de HDL. B-48 Papel estructural; necesario para el ensamblaje y secreción de los Quilomicrones. B-100 Ligando para el receptor de LDL; Papel estructural de VLDL, IDL, LDL; necesario para el ensamblaje y secreción de VLDL. CII Activa la Lipasa Lipoproteica. E Ligando para los receptores hepáticos de los remanentes de quilomicrón y de los receptores de LDL. HDL HDL Q VLDL LDL

9
Principales enzimas en el metabolismo de las lipoproteínas
Localización Función Lipoprotein Lipasa (LPL) Endotelio de los capilares del tejido muscular y adiposo Hidrólisa triglicéridos de Quilomicrón y VLDL Lecitina Colesterol Acil transferasa (LCAT) Plasma Esterifica el colesterol libre sobre la superficie del HDL Lipasa Hepática (LH) Higado Hidrolisa los triglicéridos dentro de las partículas de HDL. Proteína de transferencia de Esteres de colesterol (PTEC) Intercambia colesterol por TG con las lipoproteínas ricas en TG
 Endotelio de los capilares del tejido muscular y adiposo. Hidrólisa triglicéridos de Quilomicrón y VLDL. Lecitina Colesterol Acil transferasa. (LCAT) Plasma. Esterifica el colesterol libre sobre la superficie del HDL. Lipasa Hepática. (LH) Higado. Hidrolisa los triglicéridos dentro de las partículas de HDL. Proteína de transferencia de Esteres de colesterol. (PTEC) Intercambia colesterol por TG con las lipoproteínas ricas en TG.")
13
Fenotipo Fracción lipídica alterada Fracción lipoproteica
• I TG > 1000 mg/dl QM • IIa C total >300 mg/dl, LDL • IIb C total, TG, VLDL,LDL • III C total, TG ( mg/dl), IDL(b VLDL) • IV TG ( mg/dl), VLDL • V C total (>300 mg/dl) y TG (>1000mg/dl), VLDL, QM (+pre b lipoprot)
, IDL(b VLDL) • IV TG ( mg/dl), VLDL. • V C total (>300 mg/dl) y TG (>1000mg/dl), VLDL, QM (+pre b lipoprot)")
17
VLDL LDL IDL HDL Secreción de VLDL Lipoprotein lipasa
HIPERTRIGLICERIDEMIA Lipoprotein lipasa Síntesis apoC-III IDL Genotipo E2 Falla Poligénica HIPERLIPIDEMIA MIXTA LDL Receptor-LDL HIPERCOLESTEROLEMIA Síntesis apoA-I Expresión ABCA-1 Lipoprotein lipasa HDL COLESTEROL-HDL BAJO

28
SINDROME DE QUILOMICRONEMIA
Dentro de los defectos genéticos de la vía exógena de metabolización de las lipoproteínas, describiremos la Hipobetalipoproteinemia y Abetalipoproteinemia conjuntamente con la Enfermedad por retención de quilomicrones. Las dos primeras heredadas en forma homocigota se caracterizan por la no producción de quilomicrones, VLDL y LDL que contengan apoB. Esto ocasiona una malabsorción de grasas y una deficiencia de vitaminas liposolubles, lo que conduce a una degeneración retiniana y espinocerebelosa y a hemólisis. La abetalipoproteinemia es un trastorno autosómico recesivo infrecuente debido a un defecto en la secreción de apoB. Se ha descubierto que la anomalía genética reside en el gen que codifica una enzima microsomal de transferencia de triglicéridos. Los heterocigotas presentan una concentración normal de lipoproteínas.

29
La hipobetalipoproteinemia es causada por una anomalía del gen de la apoB.
Es un trastorno autosómico dominante relativamente raro que ocasiona una disminución de la concentración de LDL circulantes y predispone a una larga vida en los heterocigotas. La frecuencia de los heterocigotas es de cerca de 1 en 1000. La enfermedad por retención de quilomicrones (de Anderson), se caracteriza por malabsorción intestinal, ausencia de apoB-48 en la circulación y una baja concentración de LDL circulantes. Es un trastorno autosómico recesivo raro y no es causado por defecto del gen de apoB.
, se caracteriza por malabsorción intestinal, ausencia de apoB-48 en la circulación y una baja concentración de LDL circulantes. Es un trastorno autosómico recesivo raro y no es causado por defecto del gen de apoB.")
30
Estructura y función de lipoprotein-lipasa y apoC- II
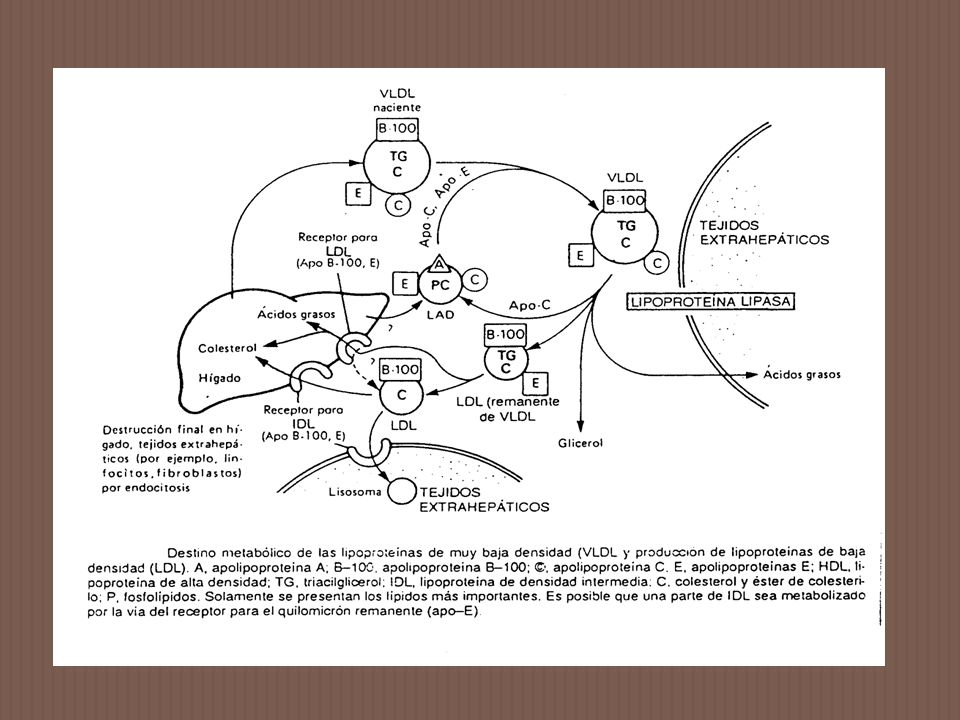
LPL es sintetizada en diferentes tejidos, pero la mayor parte se encuentra presente en tejido adiposo y músculo estriado. La enzima se sintetiza en las células parenquimales y es secretada y transportada hacia la superficie endotelial de los capilares sanguíneos, donde se une a heparán sulfato. LPL es necesaria para la eficiente hidrólisis de los TG de Q y VLDL. Para ser activa requiere de la presencia de un activador proteico apo CII sobre su superficie. Es una glicoproteína que se encuentra como un homodímero y tiene un peso molecular de daltons. Se ha clonado y secuenciado un cDNA para LPL humana que codifica para una proteína madura de 448 aminoácidos. El locus para LPL humana ha sido mapeado y ubicado en el cromosoma 8.

31
LPL es un miembro de la familia de enzimas con actividad de serina esterasas.
Un alto grado de homología existe entre el sitio catalítico de LPL y el de la lipasa hepática, lipasa pancréatica y LCAT. En tejido adiposo la actividad enzimática es inducida por insulina y aumenta en condiciones anabólicas. En músculo, la actividad permanece alta, incrementa bajo condiciones catabólicas. Estos cambios en la actividad contribuyen al almacenaje de los ácidos grasos de los TG en tejido adiposo en el estado postprandial y ayudan a proveer estos ácidos grasos a los músculos en contracción en el estado postabsortivo.

32
Apo CII es sintetizada en el hígado y secretada al plasma.
Se une a los lípidos a través de hélices anfipáticas sobre el extremo amino terminal de la proteína. Esta apoproteína recicla entre HDL y las LP ricas en TG: Q y VLDL. La porción carboxilo terminal de apo CII se une a un sitio específico de LPL lo que lleva a la activación enzimática. El gen que codifica para apo CII tiene 4 exones y 3 intrones, se encuentra en el cromosoma 19 y está íntimamente relacionado con los genes que codifican para apo E y apo CI. El gen presenta 3320 nucleótidos, mientras que el mRNA deducido del cDNA tiene solamente 494 nucleótidos de longitud.

33
Cuadro clínico En las deficiencias de LPL y apo C- II se comprueba un defecto del procesamiento de las VLDL y los quilomicrones ricos en triglicéridos. Otras características son la pancreatitis aguda y los xantomas eruptivos. El riesgo de aterosclerosis no está aumentado. Generalmente se presenta en la infancia con dolores abdominales tipo cólico, que semejan una pancreatitis, también se observa hepatomegalia. La organomegalia ocurre como resultado de la captación de los TG por los macrófagos, los que se convierten en células espumosas, esto se puede revertir con un descenso de la grasa dietaria.

34
Ya que la deficiencia de apo CII resulta en la deficiencia funcional de LPL, no es sorprendente que las manifestaciones clínicas sean similares. Aunque los síntomas aparecen a mayor edad (13-60 años). Los dos son trastornos autosómicos recesivos infrecuentes. Se han descripto varios defectos en los genes de la LPL (cromosoma 8p22) y la apoCII (cromosoma 19q13). La variación genética en estos 2 loci contribuye de manera significativa a la variación lipídica plasmática normal.
. Los dos son trastornos autosómicos recesivos infrecuentes. Se han descripto varios defectos en los genes de la LPL (cromosoma 8p22) y la apoCII (cromosoma 19q13). La variación genética en estos 2 loci contribuye de manera significativa a la variación lipídica plasmática normal.")
35
DEFICIENCIA FAMILIAR DE LA LIPOPROTEIN LIPASA
Rara. Autosómica recesiva. Un homozigoto en un millón. Niveles de Apo C-II normales. Triglicéridos altos > 1,000 mg/dl HDL bajo LDL bajo VLDL alto Quilomicrones elevados. Hígado Intestino Lipoprotein Lipasa Q rQ VLDL IDL LDL 1 2 Via endógena Via exógena

36
DIAGNOSTICO - Deficiencia de LPL
Bioquímico: En la hiperquilomicronemia se observa que los niveles de VLDL no están aumentados. Una forma de determinarlos es refrigerando el plasma por varios días; los Q flotarán en la parte superior del tubo dejando el resto del plasma claro si no hay aumento de VLDL. La ausencia de causas secundarias de hipertrigliceridemia (diabetes, alcohol, terapia estrogénica, ciertos agentes antihipertensivos) incrementa la posibilidad de una deficiencia de LPL. Un diagnóstico presuntivo se puede hacer si se produce un marcado descenso de los TG plasmáticos después de una semana de consumir una estricta dieta baja en grasas. En esta instancia también puede considerarse una deficiencia de apo CII.
 incrementa la posibilidad de una deficiencia de LPL. Un diagnóstico presuntivo se puede hacer si se produce un marcado descenso de los TG plasmáticos después de una semana de consumir una estricta dieta baja en grasas. En esta instancia también puede considerarse una deficiencia de apo CII.")
37
El diagnóstico definitivo para la deficiencia de LPL requiere del ensayo específico de determinación de la actividad de LPL en plasma post-heparina. La cosanguinidad es común en esta deficiencia. Con el desarrollo de la técnica de ELISA, para medir la inmunoreactividad de LPL en plasma, es posible demostrar que la actividad plasmática postheparina y la masa en los heterocigotas es intermedia entre los controles y los deficientes.

38
Molecular: Se ha observado que muchos pacientes parecen sintetizar LPL inmunoreactiva que no tiene actividad catalítica, por esto se debería esperar que estos enfermos presenten pequeños cambios en la secuencia de nucleótidos del gen. Con el desarrollo reciente de sondas de cDNA para LPL, el diagnóstico molecular es posible, ya que se están estudiando las distintas mutaciones. Tratamiento: -Es predominantemente por restricción de la grasa dietaria. -El objeto de esta terapia es reducir la quilomicronemia a un nivel asociado con la mejora de los síntomas y signos de este síndrome. -A menudo la restricción de un15% de las grasas es suficiente para mantener los niveles de TG por debajo de los 1000 a 2000 mg/dl.

39
DEFICIENCIA FAMILIAR DE LA APOLIPOPROTEINA C-II
Rara. Autosómica recesiva. Un homozigoto por un millón. Deficiencia o ausencia de APO C-II Hiperquilomicronemia. Los heterozigotos tienen hipertrigliceridemia leve pero no quilomicronemia ni pancreatitis. Tratamiento: Infusión de plasma normal (con Apo C-II). Hígado Intestino Lipoprotein Lipasa Q rQ VLDL IDL LDL 1 2 Via endógena Via exógena
. Hígado. Intestino. Lipoprotein Lipasa. Q. rQ. VLDL. IDL. LDL Via endógena. Via exógena.")
40
Deficiencia de apo CII -Bioquímico: esta deficiencia se manifiesta con la disminución de la actividad de LPL postheparina, en ausencia de apo CII. Con la adición de apo CII a la mezcla de reacción, aquél descenso de actividad se corrige, normalizándose. También se puede verificar por electroforesis de las apoproteínas contenidas en VLDL y Q en geles bidimensionales. -Molecular: esta deficiencia se hereda en forma autosómica recesiva. El estado heterocigota es relativamente fácil de detectar ya que los pacientes presentan la mitad de la cantidad normal de apo CII. Los heterocigotas tienen generalmente niveles plasmáticos normales de lípidos y lipoproteínas.

41
El gen de apo CII ha sido estudiado usando un grupo de enzimas de restricción (BamHI, BglI, EcoRI, HindIIIy SstI), y se observó que un polimorfismo TaqI (3,5 y 3,8 kb) existe en la vecindad de este gen. Apo CII ha sido detectada por inmunoblot o por radioinmunoensayo. Se ha observado en algunos pacientes la existencia de apo CII pero que muestra una migración anormal cuando se hizo una electroforesis bidimensional en gel. Se reportó que la región de los aminoácidos 69 al 73 sobre el extremo carboxilo terminal, relacionada con la activación de LPL, se había perdido. Tratamiento: Es el mismo que para la deficiencia de LPL.

42
HIPERTRIGLICERIDEMIA
La quilomicronemia se observa en otros estados hipertrigliceridémicos en ausencia de anormalidades en el sistema de LPL. La hipertrigliceridemia es clínicamente importante porque los niveles plasmáticos de TG por encima de 2000 mg/dl predispone a la pancreatitis. La presencia simultánea de una forma común familiar y una adquirida de hipertrigliceridemia tal como diabetes mellitus, ciertas drogas utilizadas para terapia (estrógenos) o el alcohol suelen conducir a este desorden. El tratamiento generalmente se dirige hacia ambas formas congénita y adquirida. Todas las manifestaciones clínicas del síndrome de quilomicronemia son reversibles con la reducción de los niveles de TG plasmáticos. Generalmente requiere de dietas pobres en grasas y terapias combinadas si el paciente es diabético.
 o el alcohol suelen conducir a este desorden. El tratamiento generalmente se dirige hacia ambas formas congénita y adquirida. Todas las manifestaciones clínicas del síndrome de quilomicronemia son reversibles con la reducción de los niveles de TG plasmáticos. Generalmente requiere de dietas pobres en grasas y terapias combinadas si el paciente es diabético.")
43
DEFICIENCIA DE LECITIN COLESTEROL ACIL TRASNFERESA
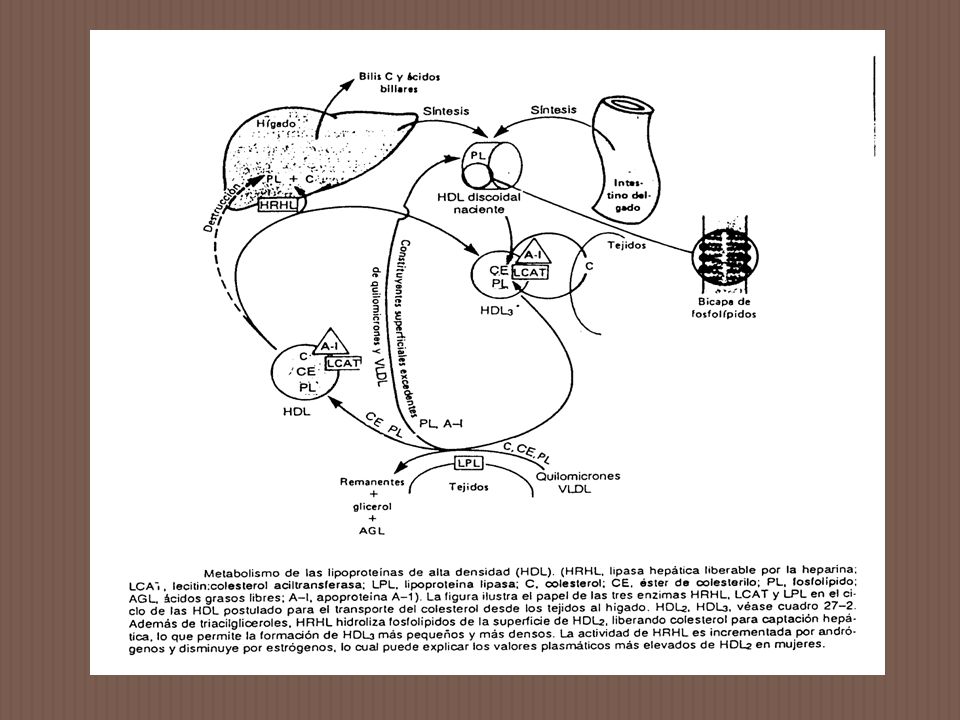
(LCAT) Características de la enzima -LCAT es una glicoproteína de aproximadamente daltons. Ha sido aislada y purificada. La estructura de esta enzima ha sido determinada por cDNA y clonado genómico, el mRNA y la secuencia de aminoácidos. La proteína madura contiene 416 aminoácidos. LCAT se sintetiza en el hígado y circula en el plasma formando complejo con HDL y la proteína que transfiere ésteres de colesterol. Es activada por apo AI. Controla indirectamente los niveles de colesterol libre y esterificado en distintas células. Cataliza la transformación de un grupo acilo de fosfatidilcolina a colesterol produciendo lisolecitina y colesterol esterificado.
 Características de la enzima. -LCAT es una glicoproteína de aproximadamente daltons. Ha sido aislada y purificada. La estructura de esta enzima ha sido determinada por cDNA y clonado genómico, el mRNA y la secuencia de aminoácidos. La proteína madura contiene 416 aminoácidos. LCAT se sintetiza en el hígado y circula en el plasma formando complejo con HDL y la proteína que transfiere ésteres de colesterol. Es activada por apo AI. Controla indirectamente los niveles de colesterol libre y esterificado en distintas células. Cataliza la transformación de un grupo acilo de fosfatidilcolina a colesterol produciendo lisolecitina y colesterol esterificado.")
44
Esta reacción se produce en la superficie de HDL.
Normalmente LCAT interviene en la maduración de HDL naciente, en la eliminación del exceso de colesterol libre de LDL, VLDL y Q. Favorece el flujo de colesterol desde las membranas celulares a HDL. La enfermedad se hereda en forma autosómica recesiva. El defecto a sido localizado en el cromosoma 16. CUADRO CLINICO Anormalidades clínicas y tisulares Las anormalidades clínicas incluyen opacidad corneal, anemia, plasma lechoso, frecuentemente proteinuria y aterosclerosis prematura. La falla renal puede ser una complicación que requiere tratamiento de por vida.

45
Dentro de las anormalidades tisulares se observan:
Células espumosas en médula ósea y riñón, e inclusiones laminares en células del bazo. La membrana del glóbulo rojo presenta aumentada concentración de colesterol libre y fosfatidilcolina. Las anormalidades en las lipoproteínas plasmáticas involucran a todas las LP y afectan la composición, distribución y concentración. La llamada Enfermedad del Ojo de pescado (Fish eye), se presenta con la deficiencia parcial en la actividad de LCAT. Caracterizada también por opacidad corneal y anormalidades en las LP plasmáticas e hipertrigliceridemia. La concentración de HDL está reducida aproximadamente en un 10% del normal y son partículas de menor tamaño.
, se presenta con la deficiencia parcial en la actividad de LCAT. Caracterizada también por opacidad corneal y anormalidades en las LP plasmáticas e hipertrigliceridemia. La concentración de HDL está reducida aproximadamente en un 10% del normal y son partículas de menor tamaño.")
46
DIAGNOSTICO BIOQUIMICO Y MOLECULAR
La actividad de LCAT en plasma se puede demostrar incubando el plasma a 37º C y midiendo la disminución de la concentración de colesterol libre plasmático. Alternativamente, colesterol libre marcado radiactivamente, introducido en LP puede ser incubado con plasma o fracciones de plasma y así se puede determinar la cantidad de ésteres de colesterol radiactivo formado. Técnicas inmunoquímicas revelan que las mutaciones conducirían a la síntesis y secreción de enzima inactiva. Como resultado de esto, se acumulan colesterol libre y fosfatidilcolina en plasma y se produce un déficit de ésteres de colesterol plasmáticos.

47
El gen de LCAT ha sido clonado, y está dividido en 6 exones.
Los datos obtenidos de la hibridización por Southern blot sugieren que hay un único gen para LCAT en el genoma humano. Los estudios sobre muestras de ADN de diferentes pacientes no relacionados revelaron que no existen grandes delecciones o rearreglos en la secuencia de este gen. Se han llevado a cabo estudios de linkage para varios sistemas marcadores genéticos. Algunos autores han usado anticuerpos monoclonales y un cDNA de LCAT para estudiar varias familias con la deficiencia. Cuando se estudió el DNA de muestras de pacientes con enzimas de restricción, se obtuvieron fragmentos cuya longitud era similar a los encontrados en los controles.

48
TRATAMIENTO Ya que hasta el presente no ha sido posible tratar la enfermedad mediante la administración de la enzima por vía intravenosa, se ha empleado transfusión de plasma o sangre total. Se ha implementado también tratamiento dietario, en particular enfocado a disminuir las LDL que aparecen como grandes partículas y relacionadas con el daño renal. Así se sugiere disminuir la ingesta de grasas. Se han debido realizar transplantes de riñón en muchos pacientes, para solucionar el síndrome nefrótico, pero no revierte las anormalidades de LP ni incrementa la actividad de LCAT. En algunos pacientes la opacidad corneal es tan intensa que es necesario llevar a cabo el transplante de córnea.

49
DEFICIENCIA DE APO E2 ApoE2: menor afinidad al receptor LDL así sus remanentes son retirados del plasma a velocidad lenta Homocigotos (E2/E2) subgrupo mas común 1% Desencadenado por: dieta rica en calorías y grasas, DM, obesidad, Hipotiroidismo, nefropatía, deficiencia de estrógeno, alcohol o hiperlipidemia combinada familiar o hipercolesterolemia familiar
 subgrupo mas común 1% Desencadenado por: dieta rica en calorías y grasas, DM, obesidad, Hipotiroidismo, nefropatía, deficiencia de estrógeno, alcohol o hiperlipidemia combinada familiar o hipercolesterolemia familiar.")
50
Rara vez antes de menopausia Xantomas Tuberoeruptivos
Adultez presentan: xantomas y cardiopatía coronaria + vasculopatía periférica prematura Rara vez antes de menopausia Xantomas Tuberoeruptivos Racimos de pequeñas pápulas en codos rodillas y nalgas = uvas XANTOMAS PALMARES: pigmentaciones blanco amarillentas en pliegues de palmas

51
TRATAMIENTO Dieta baja en colesterol y grasas
Reducir la ingesta de alcohol Reducción de peso Mujeres posmenopáusicas: inhibidores de la reductasa de HMG-CoA, fibratos y niacina

52
DIAGNOSTICO Niveles plasmáticos de col y Tg se elevan hasta que los Tg llegan a 5.6 mol/L El método tradicional diagnostico es: electroforesis de lipoproteínas (banda beta amplia) Confirma: medición de VLDL-C por ultracentrifugación además de calcular su índice (> 0.30)
 Confirma: medición de VLDL-C por ultracentrifugación además de calcular su índice (> 0.30)")
53
Xantoma Tuberoso Xantomas Eruptivos
Arco Corneal Xantelasma

54
Xantoma Tendinoso Xantoma Tendinoso Xantoma Palmar

55
HIPERLIPOPROTEINEMIAS SECUNDARIAS

Presentaciones similares