Descargar la presentación
La descarga está en progreso. Por favor, espere

1
TRASTORNOS HEMORRAGICOS CONGENITOS
Medicina II U.C.R.

2
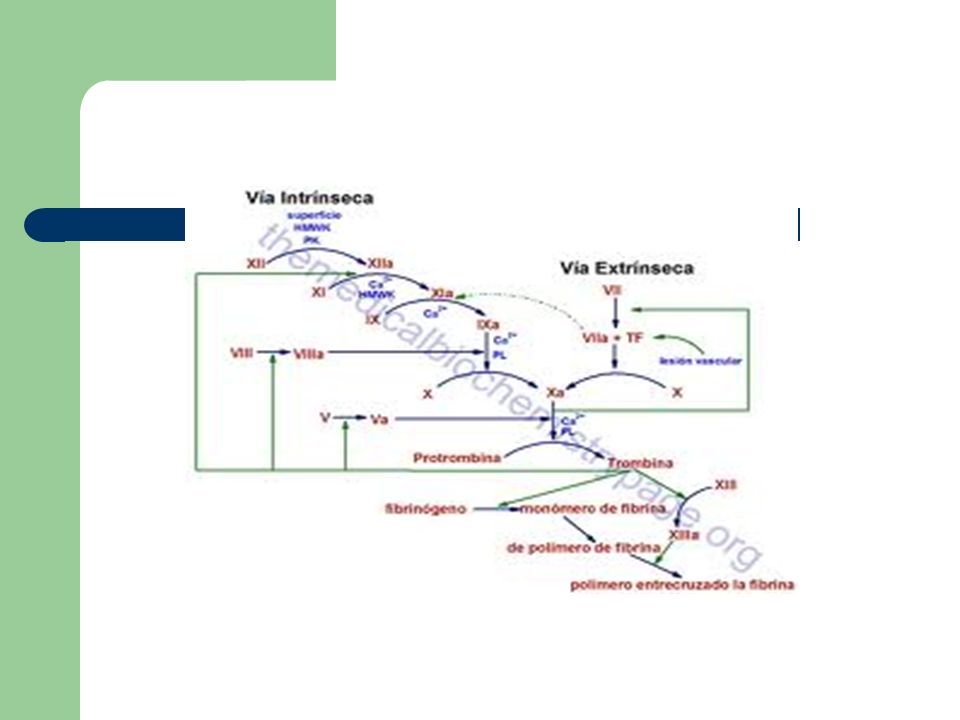
Fisiología de la coagulación
Es Sistema hemostático: Componente vascular Componente plaquetario Proteínas de la coagulación

5
Fisiología de la coagulación

7
Relevancia En el momento del diagnóstico
En el abordaje del paciente con sangrado

8
Generalidades Patrón de herencia Antecedentes hereditarios
Autosómica recesiva (vW) Antecedentes hereditarios No siempre presentes Edad a la que inician las manifestaciones Casi siempre en la infancia Patrones específicos de manifestaciones Frecuencia
 Antecedentes hereditarios. No siempre presentes. Edad a la que inician las manifestaciones. Casi siempre en la infancia. Patrones específicos de manifestaciones. Frecuencia.")
9
Enfermedad de von Willebrand
Patrón autosómico dominante Mayor frecuencia (1%) Disminución cuantitativa o por alteración cualitativa del factor vW Principales manifestaciones en piel y mucosas Diagnóstico, laboratorio
 Disminución cuantitativa o por alteración cualitativa del factor vW. Principales manifestaciones en piel y mucosas. Diagnóstico, laboratorio.")
10
Factor de von Willebrand
Funciones: Unirse a plaquetas y al subendotelio, actúa como un puente en las reacciones de la hemostasia primaria Unirse al factor VIII, evitando su proteólisis

11
Factor de von Willebrand
Glicoproteínea multimérica de gran tamaño Se sintetiza en las células del endotelio y en los megacariocitos Estructura genética localizada en el cromosoma 12

12
Manifestaciones clínicas
Frecuentes en casos moderados o severos Aparentes desde la infancia o la adolescencia Afecta por igual ambos géneros

13
Pruebas de laboratorio
Ag factor vW Actividad del factor vW (cofactor de ristocetina, unión al colágeno) Actividad factor VIII Medición de la habilidad del factor vW para formar el tapón plaquetario (PFA-100 o el tiempo de sangrado) RIPA, multímeros de vW
 Actividad factor VIII. Medición de la habilidad del factor vW para formar el tapón plaquetario (PFA-100 o el tiempo de sangrado) RIPA, multímeros de vW.")
14
Clasificación Tipo 1: Deficiencia cuantitativa parcial 70-75%
Autosómica dominante Sangrado leve o moderado Disminución en actividad del factor vW

15
Tipo 2A Autosómico dominante 10-15% Sangrado de moderado a severo
Defecto en el ensamblaje de la molécula o mayor proteólisis por ADAMTS 13 Disminución de los grandes multímeros

16
Tipo 2B Autosómico dominante 5% Moderado a severo
Mutaciones en la región de unión a la GP Ib, unión de grandes multímeros a las plaquetas Trombocitopenia

17
Tipo 2M Poco común Autosómico dominante Moderado a severo
Unión defectuosa a la GP Ib Presencia de todos los multímeros

18
Tipo 2N Disminución del nivel del factor VIII Moderado a severo
Disminución a la unión al factor VIII y menor protección a éste con disminución de su vida media Autosómico dominante

19
Tipo 3 Deficiencia severa del factor vW Rara
Herencia homocigota o doble heterocigota Manifestaciones mucocutáneas severas, en tejidos blandos y hemartrosis

20
Tratamiento Su objetivo es normalizar el valor del
F VW plasmático y así evitar los episodios hemorrágicos: Antifibrinolíticos Estrógenos DDAVP Terapia sustitutiva

21
Deficiencia de factor VII
Transmisión autosómica recesiva Cromosoma 13 Sintomática en homocigotos o heterocigotos compuestos (factor X) Síntomas variables Cursa con TP prolongado y TPTa normal
 Síntomas variables. Cursa con TP prolongado y TPTa normal.")
22
Deficiencia factor VII
El TP prolongado corrige con plasma normal Diagnóstico: Medición de los niveles de actividad del factor Diferenciar de: deficiencia adquirida del factor VII como enfermedad hepática, deficiencia vitamina K, uso de warfarina

23
Deficiencia factor X Transmisión autosómica recesiva
Sus manifestaciones se relacionan con el nivel de factor Déficit grave: <1% puede presentar sangrados espontáneos Es frecuente la historia de menorragias

24
Deficiencia Factor X Cursa con TP y TPTa prolongados
TP y TPT prolongados, con corrección Tiempo de víbora Russell prolongado Tiempo de trombina normal Diferenciar de la deficiencia de protrombina, deficiencia del factor V, deficiencias combinadas, enfermedad hepática, deficiencia vitamina K y anticoagulante lúpico

25
Deficiencia Factor XI De transmisión autosómica recesiva
Se manifiesta en homocigotos y heterocigotos compuestos Ocurre una tendencia hemorrágica de leve a moderada, dependiendo de la lesión La aparición de manifestaciones en heterocigotos puede asociarse a otras alteraciones hemostáticas

26
Deficiencia Factor XI Cursan con TP normal y TPTa prolongado
El diagnóstico se establece por estudio combinado de la coagulación con plasma deficiente en factor XI

27
Deficiencia factor XIII
Cursa con manifestaciones hemorrágicas de moderada a grave y con cicatrización alterada de las heridas Se transmite en forma autosómica recesiva Es frecuente la consanguinidad

28
Deficiencia del factor XIII
Provoca la formación de coágulos menos estables y más susceptibles a la degradación fibrinolítica Hay una tendencia hemorrágica aumentada y es frecuente el sangrado por el muñón umbilical La incidencia de sangrado intracraneal es alta

29
Deficiencia factor XIII
Cursan con TP y TPTa normales Pueden aumentar los productos de degradación de la fibrina El tiempo de trombina puede estar ligeramente prolongado Hay una solubilidad aumentada del coágulo a la solución de urea 5M

30
Tratamiento Uso de plasma fresco congelado
Uso de concentrados de factor derivados del plasma sanguíneo (Complejo Protrombínico) Crioprecipitados
 Crioprecipitados.")
31
HEMOFILIAS Deficiencias congénitas de la actividad de los factores VIII (hemofilia A) o IX (hemofilia B), de la coagulación Patrón de herencia recesiva ligada al Cromosoma X

33
Hemofilia A Hay una deficiencia en la actividad coagulantes del factor VIII Gen ubicado en el brazo largo del cromosoma X Síntesis principalmente hepática Incidencia 1:5000 varones nacidos vivos

34
Hemofilia B Deficiencia plasmática del factor IX de la coagulación, el cual es de síntesis hepática Gen localizado en el brazo largo del Cromosoma X Incidencia 1: varones nacidos vivos

35
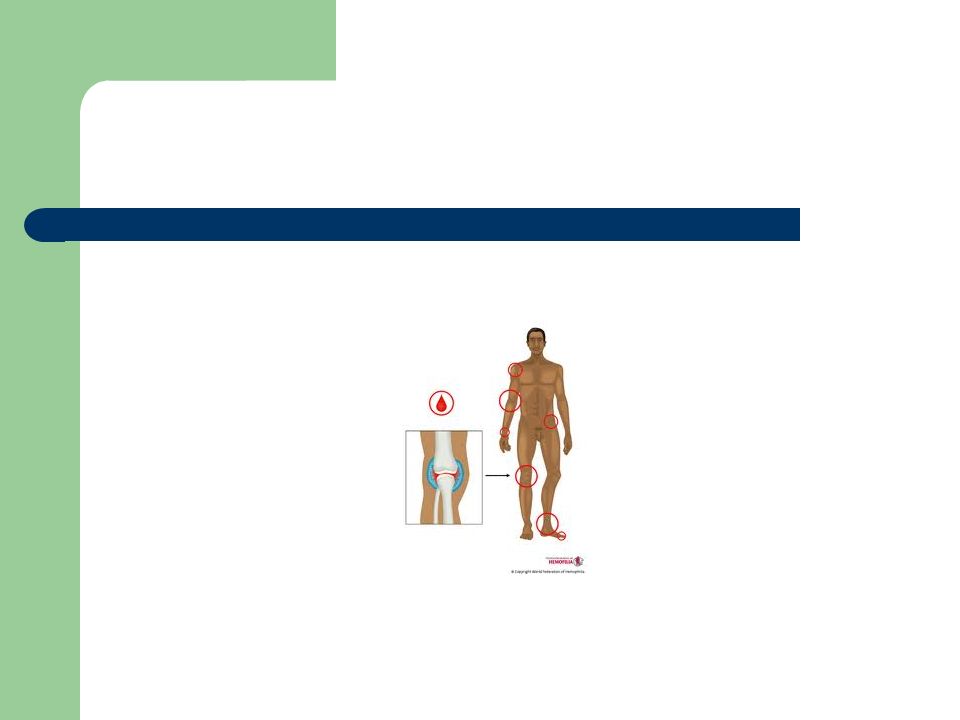
Manifestaciones clínicas
Hemorragias que aparecen por traumatismos mínimos De localización interna o externa Manifestación desde edades tempranas Complicaciones asociadas: Salud oral, artropatías, síndrome compartimental, discapacidad, hemorragias severas

43
Diagnóstico Biológico
Se basa en la dosificación de la actividad pro-coagulante correspondiente aTTP prolongado Según la dosificación de la actividad del factor se clasifican en: Leve (6-30%) Moderada (1-5%) Severas (<1%)
 Moderada (1-5%) Severas (<1%)")
44
Tratamiento Antifibrinolíticos Terapia sustitutiva
Concentrados del factor Modalidad a demanda Modalidad de profilaxis Tratamiento de soporte a nivel odontológico, ortopédico, de fisioterapia y psicológico

45
Tratamiento Complicaciones del tratamiento
Transmisión viral: Hepatitis B, Hepatitis C y VIH Aparición de Inhibidores (20%)
")
Presentaciones similares