Descargar la presentación
La descarga está en progreso. Por favor, espere

1
Coagulopatías Hemofilias Enf. Von Willebrand Púrpuras.
Trombosis venosa y arterial Dr. Oscar Mario Alvarado Rojas

2
La esplenomegalia produce trombocitosis
Plaquetas. Estructuras que carecen de núcleo miden de 2 a 4 micrómetros de diámetro. Vida media de 4 días 60 % están en torrente sanguíneo las restantes en el bazo. Sus membranas tienen receptores para colágena, ADP , factor de Von Willebrand así como fibrinógeno. Inducen a coagulación y también inducen a retracción del coágulo Contiene en su citoplasma dos tipos de gránulos Gránulos densos contienen sustancias no proteicas para la activación plaquetaria y gránulos alfa que tienen proteínas que incluyen factores de coagulación y factor de crecimiento derivado etc. Son fagocitadas por las células de Kupffer y en el bazo La esplenomegalia produce trombocitosis

3
Coagulación Este proceso es debido, en última instancia, a que una proteína soluble que normalmente se encuentra en la sangre, el fibrinógeno, experimenta un cambio químico que la convierte en insoluble ; fibrina produciendo una red que entrelaza otras moléculas y células produciendo agregados macromoleculares . Un coágulo es, por lo tanto, una red tridimensional de fibrina que eventualmente ha atrapado entre sus fibras a otras proteínas, agua, sales y hasta células sanguíneas. Por una convención se denomina "trombo" a un coágulo formado en el interior de un vaso sanguíneo.

4
Gránulos plaquetarios.
Gránulos densos: ADP , ATP, Serotonina, Calcio Gránulos alfa: factores de coagulación V y XIII, Factor V Willebrand, factor de crecimiento, fibrinogéno

6
Coagulación Las células endoteliales usualmente inhiben la activación plaquetaria por la producción de monóxido de nitrógeno, ADP asa endotelial y Prostaglandina 2. La ADP asa actúa activando la agregación en caso de secreción de ADP plaquetario El tejido endotelial libera el factor Von Willebrand en caso de lesión del tejido y este a su vez permite la adhesión de colágeno que estimula junto con el factor la activación plaquetaria.

10
Trastornos plaquetarios.
Trombocitopenia: Hemorragias Trombocitosis: Trombosis

11
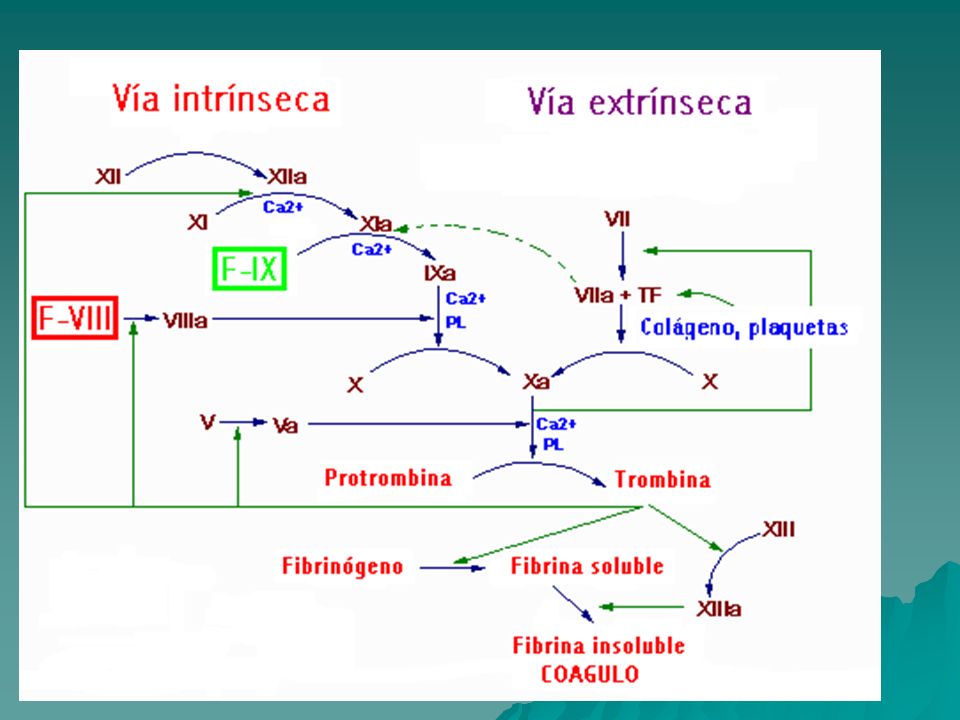
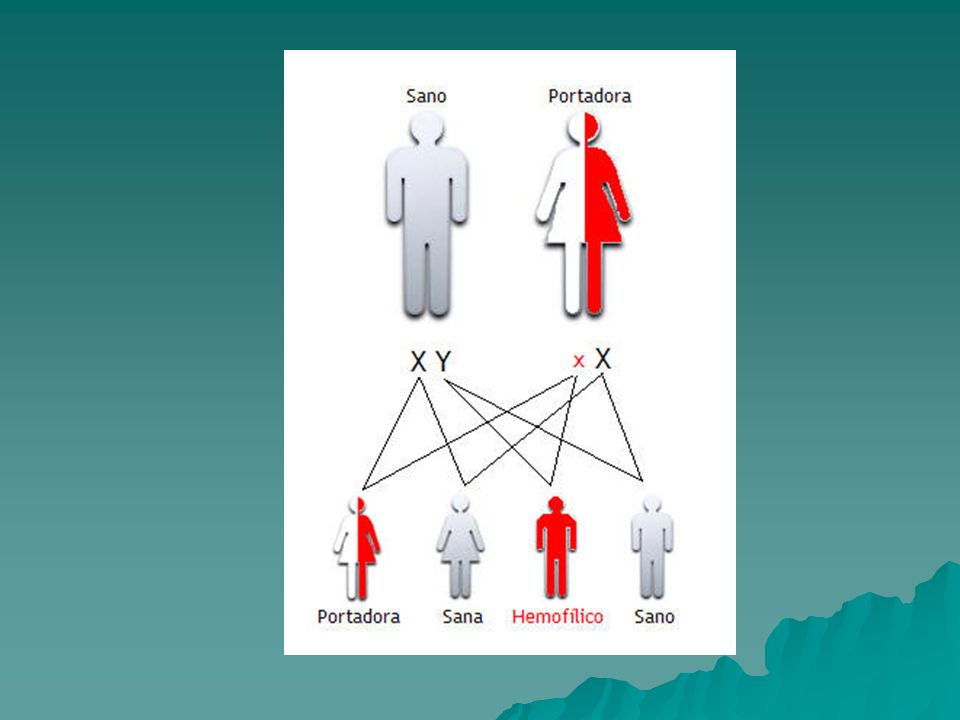
Hemofilia Enfermedad genética recesiva relacionada al sexo en el cromosoma X La hemofilia cursa con una deficiencia de los factores de la coagulación sanguínea VIII o IX que son dos eslabones clave de la cascada intrínseca de la coagulación. Así la Hemofilia A se produce porque no es del todo funcional el factor VIII y la Hemofilia B cuando no lo es el factor IX. Esto puede ser porque no hay nada de factor o bien porque el que hay no funciona adecuadamente. La frecuencia de esta enfermedad es baja por lo que a la Hemofilia se la conoce como enfermedad rara ya que, por ejemplo, la Hemofilia A se produce en 1 de cada recién nacidos vivos y la Hemofilia B en 1 de cada

13
Hemofilia

15
Enfermedad de Von Willebrand
La enfermedad de Von Willebrand es causada por una deficiencia del factor de Von Willebrand, que ayuda a las plaquetas de la sangre a aglutinarse y adherirse a las paredes de los vasos sanguíneos, lo cual es necesario para la coagulación normal de la sangre. Tiene relación directa con la ausencia de este factor ya sea en la agregación plaquetaria o bien en el transporte del factor VIII La enfermedad de Von Willebrand afecta ambos sexos puede ser heredado en forma recesiva o dominante. Usualmente es leve y se debe específicamente a trastornos en una proteína transportadora del factor VIII Se describen tres tipos. Tipo 1 Baja en la cuantía de esta proteína, Tipo 2 Mutaciones de la estructura de la proteína y tipo 3 ausencia de la proteína.

16
Púrpura Aparición de manchas violáceas en la piel
Trombocitopenia menos de ud/ul Usualmente de origen inmunológico (Púrpura trombocitopénica idiopática) Órganos inmunitarios producen anticuerpos contra las plaquetas, Esplenomegalia. PTI se observa en niños posterior a infecciones virales a la edad de 2 a 10 años. Suele ser asintomático o presentar hemorragias cutáneas persistente en áreas expuestas a los traumatismos o bien en mucosas. Son raras las hemorragias intra-articulares.
 Órganos inmunitarios producen anticuerpos contra las plaquetas, Esplenomegalia. PTI se observa en niños posterior a infecciones virales a la edad de 2 a 10 años. Suele ser asintomático o presentar hemorragias cutáneas persistente en áreas expuestas a los traumatismos o bien en mucosas. Son raras las hemorragias intra-articulares.")
17
Trombosis

18
Trombosis Puede ocasionar isquemia o infarto de tejidos.
Usualmente se localizan en endotelios vasculares deteriorados. Parciales o totales. Pueden migrar (embolia) Usualmente venosos Arterial es de aparición súbita
 Usualmente venosos. Arterial es de aparición súbita.")
Presentaciones similares














 para formar acido carbónico a partir de CO2 y agua.>")








