Descargar la presentación
La descarga está en progreso. Por favor, espere

1
Hospital Universitario Patología Clínica Dr. Rogelio Cázares Tamez.
Coagulación Hospital Universitario Patología Clínica Dr. Rogelio Cázares Tamez.

2
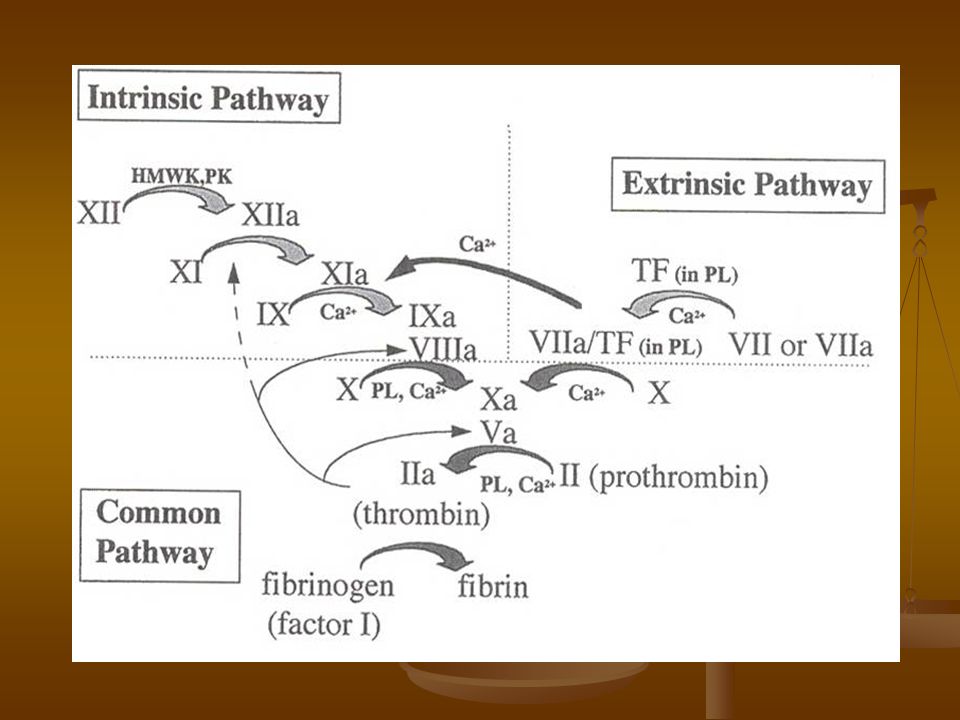
Cascada de la Coagulación
Históricamente se han descrito 2 vías: - Intrínseca: inicia con el factor XII, XI, IX, VIII, Precalicreína y CAPM - Extrínseca: inicia con el factor VII y Factor Tisular. - Común: activación del factor X y es donde convergen ambas vías.

4
Fibrinolisis Se encarga de eliminar el trombo que ya no es necesario. Se inicia por TPA y UPA. Convierten el plasminógeno en plasmina. Actúa sobre el fibrinógeno y fibrina (PDF y dímeros D) Inactiva a los factores V y VIII. El factor XII activa este sistema Este es proceso es regulado por la antiplasmina y Inhibidor TPA.
 Inactiva a los factores V y VIII. El factor XII activa este sistema. Este es proceso es regulado por la antiplasmina y Inhibidor TPA.")
6
Mediciones de Laboratorio
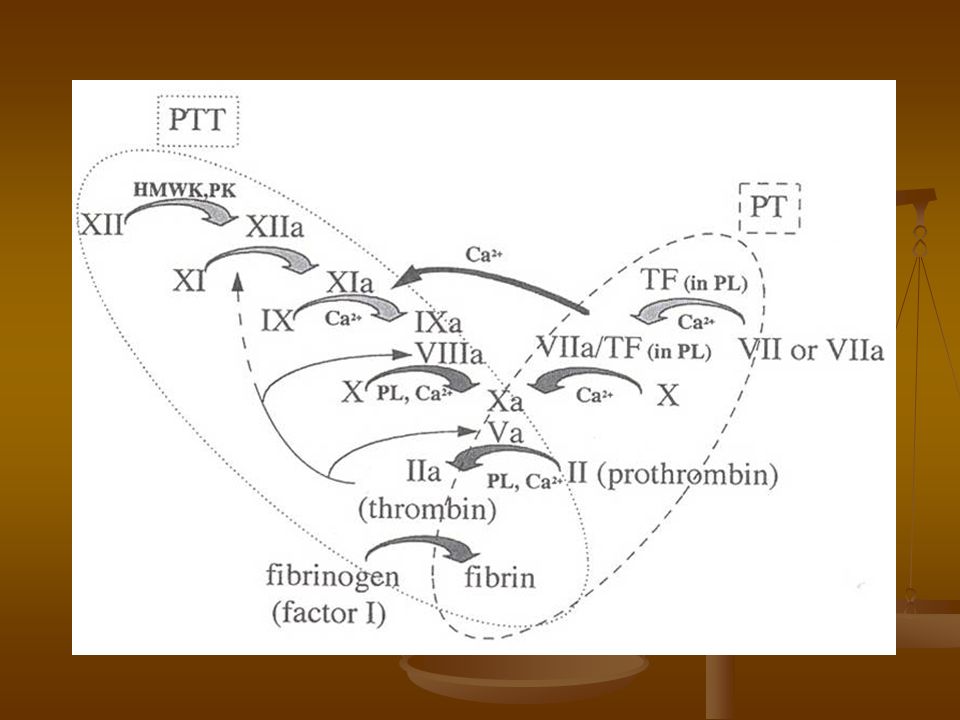
TTP: mide el tiempo de formación del trombo de fibrina desde la activación del factor XII. (Vía Intrínseca). Se requiere: Ca Fosfolípidos Activador de la Vía Intrínseca (Caolín, Sílica, Ac. Elágico).
. Se requiere: Ca. Fosfolípidos. Activador de la Vía Intrínseca (Caolín, Sílica, Ac. Elágico).")
7
Mediciones de Laboratorio
TP: mide el tiempo de formación del trombo de fibrina a partir de la activación del factor VII. (Vía Extrínseca). Se requiere: Ca Tromboplastina (fosfolípidos + FT)
. Se requiere: Ca. Tromboplastina (fosfolípidos + FT)")
9
Deficiencias de factores I, II, V, X (vía común): alargan TP y TTP.
Deficiencias de factores VII, II, V y X: alargan TP sin alterar TTP. Deficiencia de factores VIII, IX, XI XII: alargan únicamente el TTP.

11
Trastornos de la Coagulación Sanguínea
Historia clínica (AHF) Características de los sangrados Establecer si el defecto es adquirido o congénito Establecer si el defecto es en factores o en plaquetas.
 Características de los sangrados. Establecer si el defecto es adquirido o congénito. Establecer si el defecto es en factores o en plaquetas.")
12
Trastornos Hereditarios de la Hemostasia

13
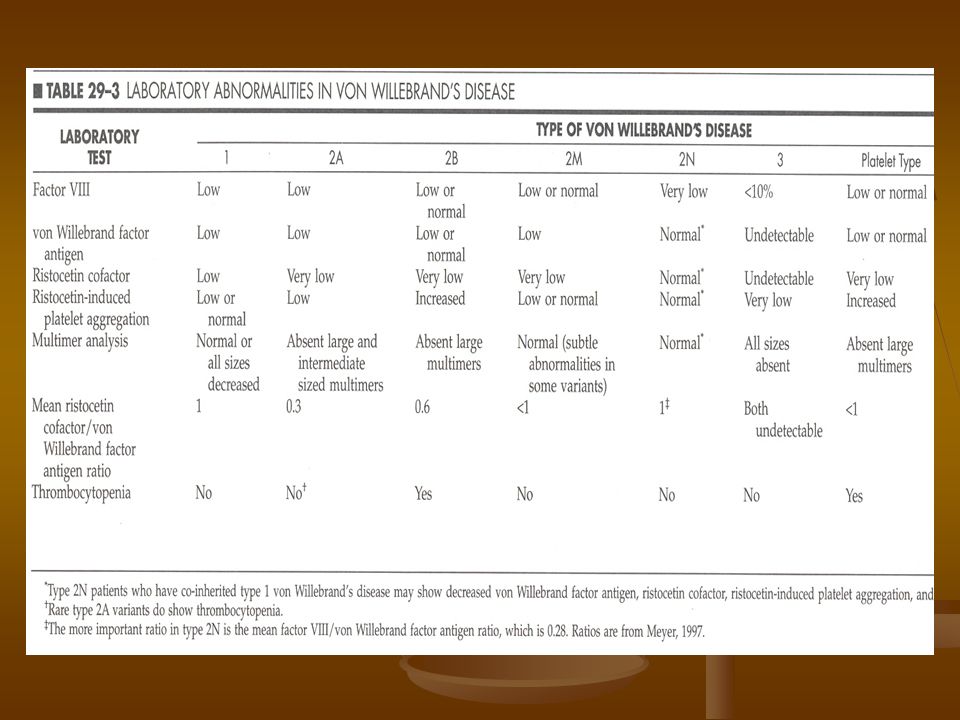
Enfermedad de Von Willebrand
Es la mas común de este grupo (1% de la población) El FvW es sintetizado por el endotelio y megacariocitos Induce agregación por medio de la GPIb, presente a la superficie de las plaquetas Se une al factor VIII alargando su vida media
 El FvW es sintetizado por el endotelio y megacariocitos. Induce agregación por medio de la GPIb, presente a la superficie de las plaquetas. Se une al factor VIII alargando su vida media.")
14
Manifestaciones clínicas
Epistaxis Gingivorragias Petequias Equimosis Sangrado abundante en procedimientos dentales.

16
Pruebas para FvW Agregometría mediada por Ristocetina
ELISA: mide cantidad pero no calidad F VIII: indirectamente midiendo el TTP

17
Hemofilia Es la enfermedad hemorrágica severa más común
Prevalencia de 1:5000 en el tipo A y de 1:25000 en el tipo B Es ligada al X afectando a varones y siendo la mujeres portadoras, salvo otras alteraciones cromosómicas ligadas al X.

18
Se divide en tres tipos:
Severa: con menos de 1% de actividad, con sangrados espontáneos Moderada: actividad del 1-5% sangrados moderados Leve: actividad en 5%. Sangrados secundarios a traumatismo o Qx.

19
Cuadro Clínico Hemartrosis, hematomas en tejidos blandos, epistaxis, sangrados abundantes en procesos quirúrgicos o dentales, hemorragia intracraneana Característicamente TTP prolongado (actividad del 20-30%) TP normal y plaquetas normales La relación FVIII-FvW en .5-1 El F VIII es lábil a temperatura ambiente, un mal manejo de la muestra puede alterar el resultado
 TP normal y plaquetas normales. La relación FVIII-FvW en El F VIII es lábil a temperatura ambiente, un mal manejo de la muestra puede alterar el resultado.")
20
Deficiencia Hereditarias de Otros Factores
Las deficiencias de F I, II, V, VII, X, XII, precalicreína y CAPM, son raras Disfribrinogenemia es una alteración en la calidad, asociada mas a trombosis que a sangrados, con TT y Tiempo de reptilasa alargados Las asociadas a F XII, precalicreína y CAPM no se relacionan con riesgo de sangrado

21
Trastornos Adquiridos de la Hemostasia

22
Coagulación Intravascular Diseminada
Estimulación exagerada del sistema de coagulación, por daño tisular extenso, quemaduras, sepsis Los sistemas de fibrinolíticos y de anticoagulación no pueden contener al estimulación, formándose, microtrombos a nivel sistémico Plaquetas, factores de coagulación y anticoagulantes son depletados, esto aumenta el riesgo de sangrado

23
En la fase aguda el TTP esta alargado en un 50% de los casos, TP 70% y Fibrinógeno disminuido en 50%
PDF y Dímeros D elevados. Este último característico de la fase aguda En la fase crónica el organismo es capaz de compensar por lo que se observan plaquetas y fibrinógeno aumentados y los tiempos se acortan En FSP se observan esquistocitos .

24
Insuficiencia hepática
Afecta principalmente al factor VII por su vida media corta Otros factores no se ven afectados El TP se prolonga primero que el TTP Coexiste con una disfibrinogenemia

25
Terapia con Anticoagulantes

26
Anticoagulantes Orales
Warfarina Actúa dañando la producción de vitamina K activa Esta intervienen en la descarboxilación de los factores II, VII, IX proteína C y S, para que estos factores sean activos Alarga el TP La vitamina K se administra en casos de sobredosis de warfarina, observado su efecto en hrs Se monitoriza con el INR

27
Anticoagulantes Intravenosos
Heparina Inhibe a los factores II, IX, X, XI, XII Alarga además el TTP En caso de sobredosis se utiliza la protamina Se recomienda el monitoreo de plaquetas cada 20 días durante el uso prolongado de heparina Se ha descrito la trombocitopenia asociada a Heparina

28
Vías Normales de Anticoagulación

29
Los sistemas principales son:
Proteína C y Proteína S Antitrombina y Factor Tisular del Plasminógeno 1 Este es activado por la Trombina y Trombomodulina Finalmente la Proteína C activada degrada al factor V y VIII, además de promover la fibrinolisis La antitrombina funciona como anticoagulante inhibiendo la trombina y a los factores X, IX, XI, XII, calicreína

30
Estados Hereditarios de Hipercoagulabilidad

31
Resistencia a Proteína C Activada
Es la causa mas común La proteína C inactiva al factor V y VIII eliminado un residuo específico de arginina Pacientes con una mutación del factor V presentan resistencia a este proceso Incrementa el riesgo de trombosis hasta 3 o 7 veces en heterocigotos y 80 en homocigotos

32
La prueba para detectar esta resistencia consiste en agregar proteína C activada exógena al suero problema y ver cuanto se alarga el TTP y se establece un rango Si este es menor a 2 indica resistencia El Gold Standard análisis de DNA por PCR

33
Deficiencia de Proteína C, Proteína S y Antitrombina
Representan el 1-9% de los casos de trombosis venosa La deficiencia de antitrombina implica mayor riesgo de trombosis que las demás deficiencia y es mas asociada a trombosis arterial Los defectos severos en RN inducen CID y púrpura fulminans La deficiencia en homocigotos es grave y sin Tx es incompatible con la vida

34
Otras alteraciones Mutación del Gen de Protombina Hiperhomocisteinemia
Disfibrinogenemia Anormalidades en trombomodulina y TFPI Defectos de la fibrinolisis

35
Estados de Hipercoagulabilidad Adquirida

36
Anticuerpos Antifosfolípidos
Son anticuerpos dirigidos contra complejos fosfolípidos-proteínas Están asociados a un riesgo elevado de trombosis arterial o venosa y trombocitopenia Los 2 principales sin anticoagulante lúpico y anticuerpo anticardiolipina.

37
In Vitro producen anticoagulación, sin embargo In Vivo inducen trombosis
Alteran todas las pruebas de laboratorio dependiente de fosfolípidos (TTP, TP, TVVR). La fisiopatogenia no se conoce bien Se propone una resistencia al proteína C, proteína S, trombomodulina Más recientemente una alteración directa sobre la beta 2 GPI (anticoagulante antiagregante).
. La fisiopatogenia no se conoce bien. Se propone una resistencia al proteína C, proteína S, trombomodulina. Más recientemente una alteración directa sobre la beta 2 GPI (anticoagulante antiagregante).")
38
Otras alteraciones: Anticonceptivos orales Embarazo Cirugía ortopédica

Presentaciones similares




















 es una alteración fisiopatólogica sistémica, trombohemorrágica, que se presenta en algunas situaciones.>")
