Descargar la presentación
La descarga está en progreso. Por favor, espere

1
GENETICA y TECNICAS DE BIOLOGIA MOLECULAR Química Biológica Patológica Dra. Silvia Varas qbpatologica.unsl@gmail.com Tema:1 (5) V- Parte 2015
 V- Parte")
2
Resumen Concepto Epidemiología Patogenia. Bases genéticas Manifestaciones clínicas generales Diagnóstico individual y poblacional Prevención. Programas de Pesquisa neonatal Bases del tratamiento

3
Errores congénitos del metabolismo

4
Edad Neonatal Lactante Niño mayor Adolescente Adulto Circunstancias desencadenantes Nutrientes Enfermedad intercurrente Fármacos Síntomas Crónico progresivo Agudo intermitente Agudo progresivo Errores congénitos del metabolismo: Presentación Clínica

5
Errores congénitos del metabolismo Categoría 1 1. Son las enfermedades que afectan 1 sistema funcional o 1 órgano 2. Presenta síntomas uniformes Categoría 2 1. Son las enfermedades que afectan una vía metabólica expresada en una gran cantidad de órganos o células. 2. Presentan síntomas muy diversos. Y se dividen en 3 grupos Grupos: 1. Síntesis/Deg moléculas complejas. 2. Afectan el metabolismo intermedio. 3. Defectos que generan alteraciones en la energía

6
Categoría 2 Grupo 1: Enfermedades que afectan la síntesis o catabolismo de moléculas complejas (disturbios en tráfico intracelular, lisosomas, etc). No corrigen con dieta. Grupo 2: Enfermedades que afectan el metabolismo intermedio. Con un aguda o progresiva intoxicación por acumulación de un metabolito por bloqueo de una vía. (Ej: aminoácidurias, intolerancia a azucares, etc). Diagnóstico: cuantificación de metabolito en plasma u orina. Tratamiento: remoción toxinas. Grupo 3: Desordenes con deficiencia de la Energía. Hay una deficiencia en la producción de energía en hígado, miocardio, músculo o cerebro. Ej: glucogenosis, defectos en la beta oxidación de ácidos grasos, etc. Hay una acumulación de síntomas y manifestaciones. Síntomas comunes: hipoglucemia, hiperlactacidemia, hipotonía, miopatía, cardiomiopatía, retraso en el desarrollo, colapso circulatorio, etc
. Diagnóstico: cuantificación de metabolito en plasma u orina. Tratamiento: remoción toxinas. Grupo 3: Desordenes con deficiencia de la Energía. Hay una deficiencia en la producción de energía en hígado, miocardio, músculo o cerebro. Ej: glucogenosis, defectos en la beta oxidación de ácidos grasos, etc. Hay una acumulación de síntomas y manifestaciones. Síntomas comunes: hipoglucemia, hiperlactacidemia, hipotonía, miopatía, cardiomiopatía, retraso en el desarrollo, colapso circulatorio, etc.")
7
Errores congénitos del metabolismo: Síndromes característicos Enfermedad Metabólica Aguda del RN (1) Síndrome Neurológico (2) Síntomas Oculares (3) Síntomas en Piel (4) Síntomas Hematológicos (5) Síntomas o Síndromes Viscerales (Intestino, Hígado, Corazón, Pulmón, Riñón) (6) Síntomas Endocrinos (7)
 Síndrome Neurológico (2) Síntomas Oculares (3) Síntomas en Piel (4) Síntomas Hematológicos (5) Síntomas o Síndromes Viscerales (Intestino, Hígado, Corazón, Pulmón, Riñón) (6) Síntomas Endocrinos (7)")
8
Enfermedad metabólica aguda del RN (1) Dificultades de alimentación Escasa ganancia ponderal Hipotonía Convulsiones Letargia Olor peculiar Hipoglucemia, hiperamoniemia, Acidosis láctica RN con Deterioro neurológico Presentación predominantemente hepática Hipoglucemia con hepatomegalia Retraso en el crecimiento intrauterino Ictericia colestática Enfermedad metabólica aguda (ECM) Exámenes de Laboratorio Glucemia Amonemia pH y bicarbonato Calcio, Magnesio Cuerpos cetónicos, AGL Acido Láctico Gases en sangre
 Dificultades de alimentación Escasa ganancia ponderal Hipotonía Convulsiones Letargia Olor peculiar Hipoglucemia, hiperamoniemia, Acidosis láctica RN con Deterioro neurológico Presentación predominantemente hepática Hipoglucemia con hepatomegalia Retraso en el crecimiento intrauterino Ictericia colestática Enfermedad metabólica aguda (ECM) Exámenes de Laboratorio Glucemia Amonemia pH y bicarbonato Calcio, Magnesio Cuerpos cetónicos, AGL Acido Láctico Gases en sangre")
9
Síndromes clínicos (2) Presentación aguda de síntomas Acidosis metabólica Cetosis Hiperlactacidemias Hipoglucemia Ataques recurrentes de coma, vómitos, letargia ataxia y síntomas psiquiátricos Dolor abdominal
 Presentación aguda de síntomas Acidosis metabólica Cetosis Hiperlactacidemias Hipoglucemia Ataques recurrentes de coma, vómitos, letargia ataxia y síntomas psiquiátricos Dolor abdominal")
10
Síndromes clínicos (2) Sintomas Neurológicos Progresivos Progresiva deterioro neurológico y mental: Infancia temprana, Infancia tardía, adolescencia o adultez: Encefalopatía aguda (hiperamoniemias, hiperglicinemias) Encefalopatía crónica (PKU, esfingolipidosis) Signos extrapiramidales: alteraciones del movimiento (distonía, ataxia) Hipotonía en periodo neonatal/Parálisis (miopatía mitocondrial, miastenia cong.)
 Sintomas Neurológicos Progresivos Progresiva deterioro neurológico y mental: Infancia temprana, Infancia tardía, adolescencia o adultez: Encefalopatía aguda (hiperamoniemias, hiperglicinemias) Encefalopatía crónica (PKU, esfingolipidosis) Signos extrapiramidales: alteraciones del movimiento (distonía, ataxia) Hipotonía en periodo neonatal/Parálisis (miopatía mitocondrial, miastenia cong.)")
11
Síndromes clínicos (3) Síntomas Oculares: Cataratas Opacidad Corneal Retinitis pigmentosa Punto Rojo Macular
 Síntomas Oculares: Cataratas Opacidad Corneal Retinitis pigmentosa Punto Rojo Macular")
12
Síndrome de depósito Enfermedades lisosomales Mancha Roja Macular

13
Síndromes clínicos (4) Síntomas en Piel: Fascies caracteristicas Lesiones en Piel Angioqueratoma
 Síntomas en Piel: Fascies caracteristicas Lesiones en Piel Angioqueratoma")
14
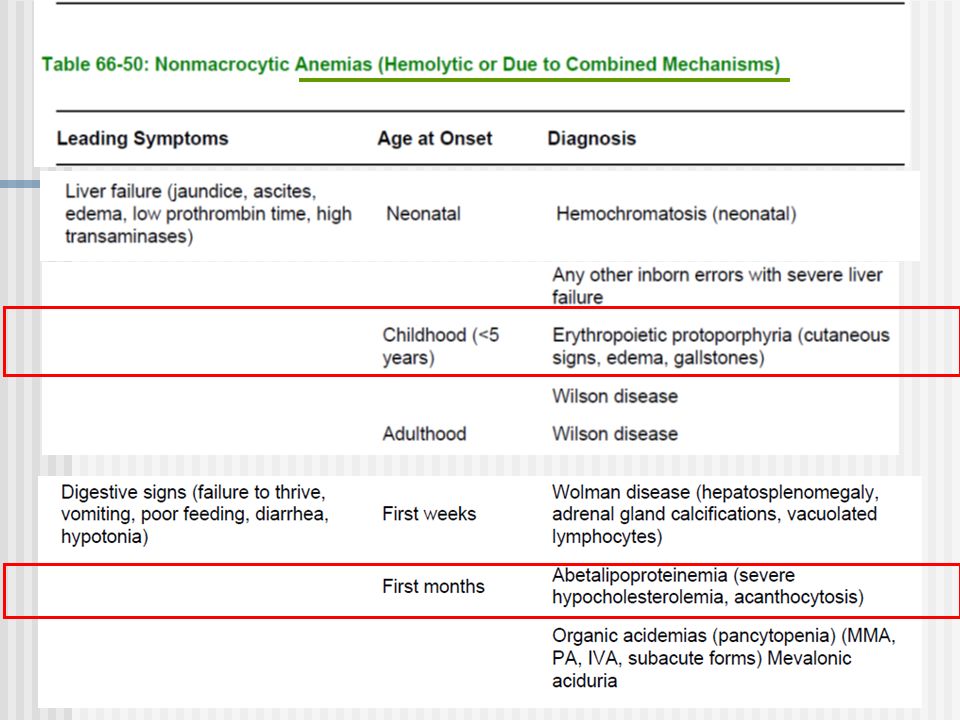
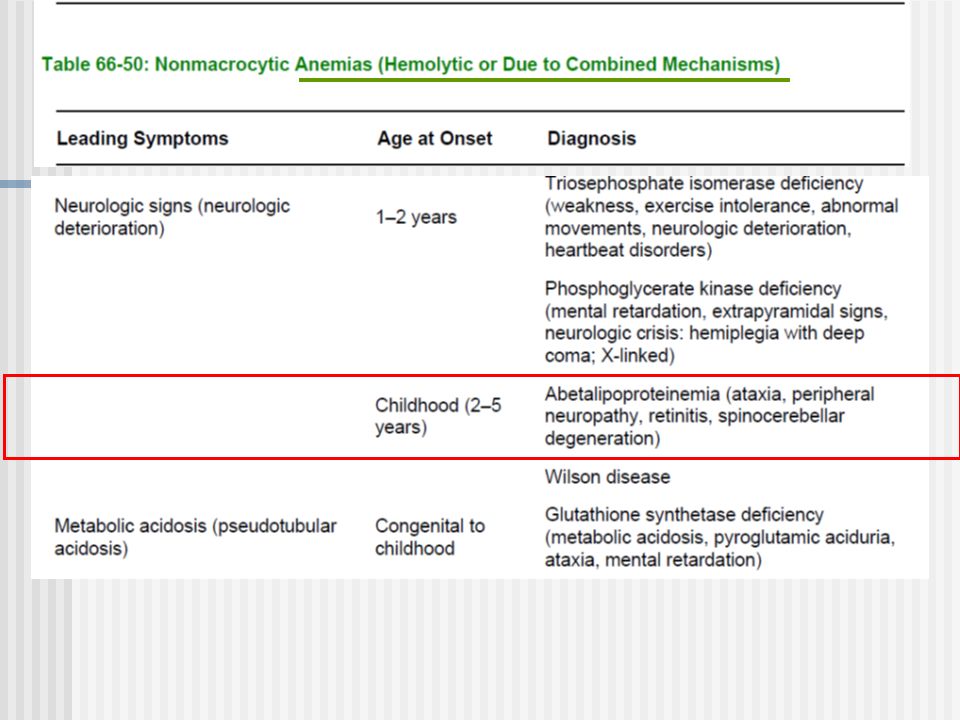
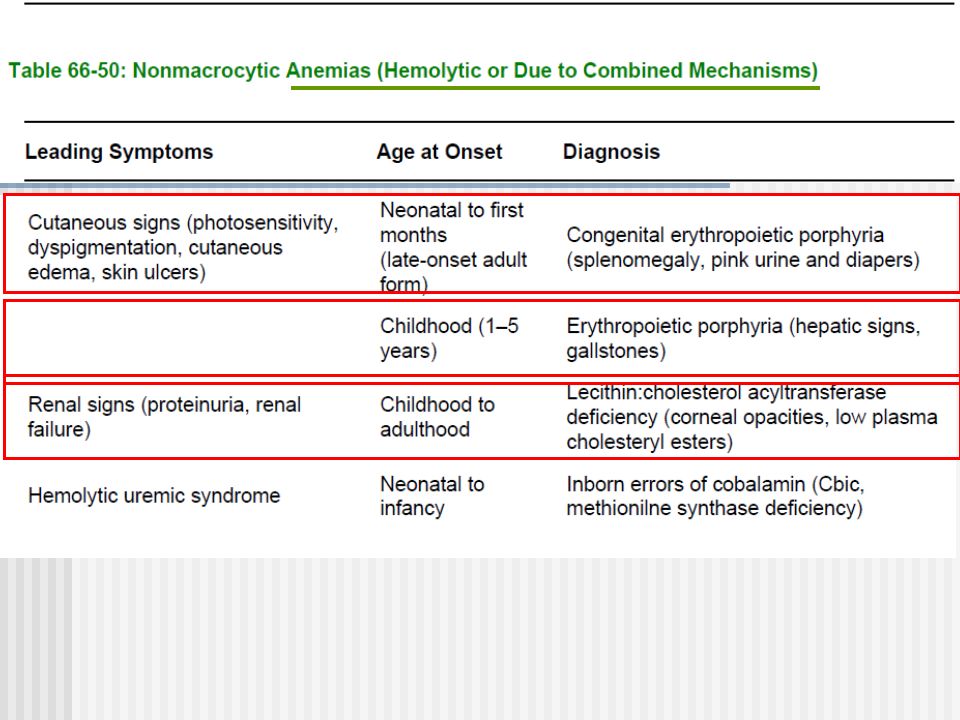
Síndromes clínicos (5) Síntomas Hematológicos Anemias no regenerativas (macrociticas) Anemias hemolíticas Pancitopenia, neutropenia, trombocitopenias Acantocitois
 Síntomas Hematológicos Anemias no regenerativas (macrociticas) Anemias hemolíticas Pancitopenia, neutropenia, trombocitopenias Acantocitois")
15
Manifestaciones clínicas (6)
")
19
Síndromes clínicos (6) Síndrome Viscerales(intestino, hígado, corazón, pulmón y riñón) Cardiomiopatía, Arritmias, Enfermedad coronaria y arterioesclerosis, Xantomas Diarreas crónicas Falla hepática
 Síndrome Viscerales(intestino, hígado, corazón, pulmón y riñón) Cardiomiopatía, Arritmias, Enfermedad coronaria y arterioesclerosis, Xantomas Diarreas crónicas Falla hepática")
20
Tratamiento en la fase aguda 1.Medidas generales de soporte Balance hidroelectrolítico Aporte de glucosa 2.Depuración de toxinas Forzar diuresis, alcalinización orina, etc Quelantes del amonio (benzoato, fenilacetato, fenilbutirato) Detoxificantes de ácidos orgánicos (glicina) Depuración extrarrenal: diálisis peritoneal, hemofiltración, exanguino-transfusión 3.Aporte de factores deficitarios Aminoácidos (arginina) Vitaminas (B12, biotina) Carnitina Apo CII
 Detoxificantes de ácidos orgánicos (glicina) Depuración extrarrenal: diálisis peritoneal, hemofiltración, exanguino-transfusión 3.Aporte de factores deficitarios Aminoácidos (arginina) Vitaminas (B12, biotina) Carnitina Apo CII")
21
Tratamiento 1.Tratamiento del fenotipo clínico Educación sanitaria Cirugía / Ortopedia Tratamiento farmacológico Trasplante de órganos

22
Tratamiento 2.Tratamiento del fenotipo bioquímico Restricción de la ingesta del sustrato (dietética) Inhibición de la síntesis del metabolito o del sustrato (quelantes) Bloqueo de receptores celulares Uso de vías metabólicas alternativas (dosis farmacológicas de vitaminas) Remplazamiento del producto deficiente (tratamiento sustitutivo)
 Inhibición de la síntesis del metabolito o del sustrato (quelantes) Bloqueo de receptores celulares Uso de vías metabólicas alternativas (dosis farmacológicas de vitaminas) Remplazamiento del producto deficiente (tratamiento sustitutivo)")
23
Tratamiento 3.Sustitución de la proteína/enzima anormal o disfuncional Trasplante de órgano (tirosinemia) Remplazamiento enzimático (Gaucher, Pompe) Ingeniería genética (vectores con DNA modificado: distrofia muscular, déficit LPL) Trasplante de progenitories hematopoyéticos: médula ósea, cordón umbilical Trasplante de stem cell (células madre)
 Remplazamiento enzimático (Gaucher, Pompe) Ingeniería genética (vectores con DNA modificado: distrofia muscular, déficit LPL) Trasplante de progenitories hematopoyéticos: médula ósea, cordón umbilical Trasplante de stem cell (células madre)")
24
Tratamiento

25
Prevención Consejo genético Requiere el diagnóstico exacto del trastorno y conocer su pronóstico (mortalidad, morbilidad) Patrón de herencia conocido (riesgo en otro descendiente) Screening Cascada Condicionamientos éticos de los padres Sobre el embrión ( FIV, Fertilización “in vitro”) ) Biopsia vellocidades coriónicas Amniocentesis
 Patrón de herencia conocido (riesgo en otro descendiente) Screening Cascada Condicionamientos éticos de los padres Sobre el embrión ( FIV, Fertilización in vitro ) ) Biopsia vellocidades coriónicas Amniocentesis")
26
Diagnóstico Diagnóstico prenatal Pre-implantación (fertilización in vitro) Amniocentesis (líquido amniótico) y biopsia fetal (>16 semanas) Biopsia vellocidades coriónicas (embrión implantado)
 Amniocentesis (líquido amniótico) y biopsia fetal (>16 semanas) Biopsia vellocidades coriónicas (embrión implantado)")
27
Diagnóstico I Diagnóstico individual - Presentación y manifestaciones clínicas - Laboratorio (inicial) Sangre: Hematología, ionograma, glucosa, Ca, P, Mg 2+, láctico/pirúvico, amonio, aminoácidos, carnitina, ácidos orgánicos, fenilpiruvico. Orina: Cuerpos cetónicos, Azúcares reductores, Ac. orgánicos LCR: proteínas, lactato y lactato/pirúvico

28
Diagnóstico II - Laboratorio avanzado Sangre: Enzimas, ácidos orgánicos, carnitina, Ácidos Grasos Libres, niveles de Hormonas basales y post-estimulación, sobrecarga KClO 4. - Biología molecular (DNA GENÓMICO): análisis de mutaciones - Biopsias: histoquímica, enzimología, - Imagen: Ecografía, TAC, etc.
: análisis de mutaciones - Biopsias: histoquímica, enzimología, - Imagen: Ecografía, TAC, etc..")
29
Pesquisa neonatal sistemático en recién nacidos (criterios OMS) Programa integrado con recursos legales, humanos y materiales. Educación de la población y del personal sanitario. Estandarización del test de “screening”, que debe ser: - Método simple. - Relación coste/beneficio adecuada. - Alta sensibilidad (% de niños afectados con test +) - Alta especificidad (% de niños sanos con test -). Recuperación de los niños con resultado positivo. Confirmación diagnóstica. Tratamiento. Seguimiento y evaluación de los pacientes
 - Alta especificidad (% de niños sanos con test -). Recuperación de los niños con resultado positivo. Confirmación diagnóstica. Tratamiento. Seguimiento y evaluación de los pacientes .")
30
Pesquisa Neonatal en Argentina - 1986: La pesquisa neonatal de la fenilcetonuria y el hipotiroidismo congénito es obligatoria en nuestro país por la ley 23.413 y 23.874, respectivamente. - 1995: se incorporo la Pesquisa de Fibrosis Quística a todos los recién nacidos por la ley 24.438.

31
LEYES PROVINCIALES SL En la Provincia de San Luis rigen la Ley N° III- 00767-2004 (5436) que establece la obligatoriedad de detección de Hipotiroidismo Congénito, Fenilcetonuria y Fibrosis Quística, y la Ley N° III-0077-2004 (5435) que establece la obligatoriedad para la prevención de Galactosemia e Hiperplasia Suprarrenal Congénita.
 que establece la obligatoriedad de detección de Hipotiroidismo Congénito, Fenilcetonuria y Fibrosis Quística, y la Ley N° III (5435) que establece la obligatoriedad para la prevención de Galactosemia e Hiperplasia Suprarrenal Congénita.")
32
Pesquisa Neonatal - Muestra de sangre desecada - Todos los recién nacidos a partir de las 24 hs y hasta el 7º día de vida.

33
Al 2013 es obligatorio la Pesquisa de: Hipotiroidismo congénito Fenilcetonuria Fibrosis Cística Galactosemia Hiperplasia Suprarrenal Congénita Deficiencia de Biotinidasa

34
Material de extracción lanceta Aguja metálica nº 25G mariposa nº 27G Papel Cromatográfico

35
Sector donde debe hacerse la punción en el pie del neonato

36
Zona de extracción con (lanceta)
")
37
Talón (lanceta) Dorso de la Mano
 Dorso de la Mano")
38
Calidad de las muestras de sangre obtenidas OPTIMO

40
Epidemiología Bs As PKU, FENILCETONURIA 1/10.000 Hipotiroidismo 1/2.500 Fibrosis Cística 1/6.500 Enfermedad Incidencia

42
Datos del grupo de CS-SL: Dra Adriana Pastrán y col. Gracias!

43
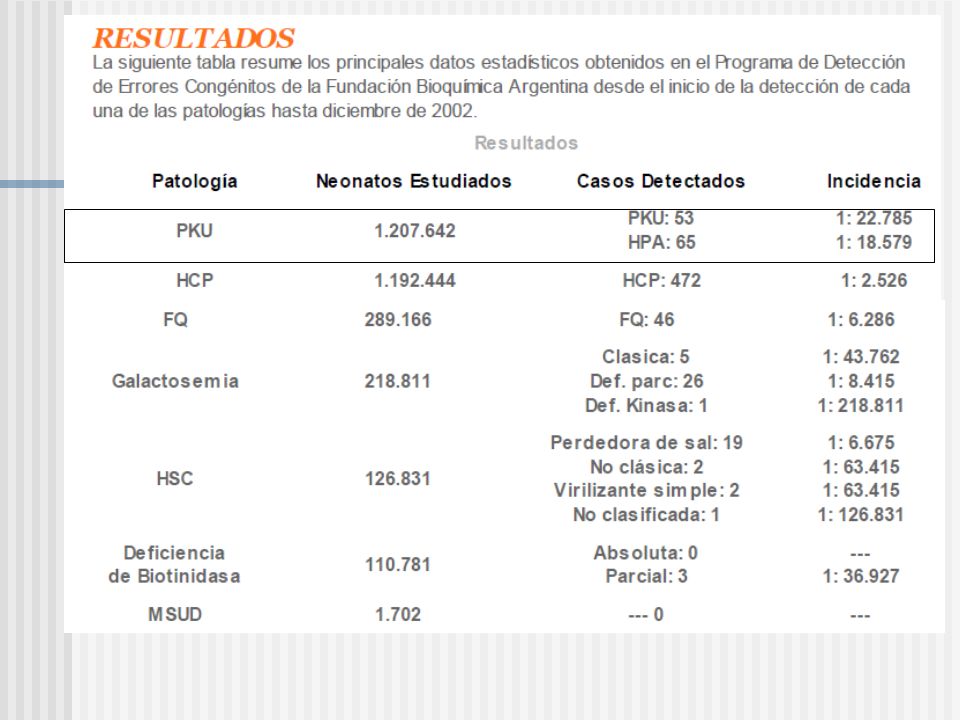
Según el Manual del MSN: - Hipotiroidismo Congénito Primario (HCP) 1:2.500 recién nacidos. - Fibrosis quística Frecuencia 1: 2.500 nacidos vivos - Fenilcetonuria 1:10.000 a 1:20.000 nacidos vivos - Hiperplasia Suprarrenal Congénita (HSC) La frecuencia de 1:12.000 nacidos vivos. - Deficiencia de Biotinidasa La frecuencia de esta enfermedad es de 1:45.000. - Galactosemia La frecuencia es de 1:60.000 nacidos vivos 9,5 casos en 23.600 casos 1-2 casos en 23.600 casos 2 casos en 23.600 casos 9,5 casos en 23.600 casos o 3 casos en 23.600 casos
 La frecuencia de 1: nacidos vivos. - Deficiencia de Biotinidasa La frecuencia de esta enfermedad es de 1: Galactosemia La frecuencia es de 1: nacidos vivos 9,5 casos en casos 1-2 casos en casos 2 casos en casos 9,5 casos en casos o 3 casos en casos.")
44
AÑO TOTAL ANALIZA- DOS HC (+)PKU (+)TIR(+)17-OH Pg (+)GALACTOSA BIOTINI DASA 2009 (sep- dic)1321000000 201042855 (*)00100 20114377303 (* *)000 20124627101000 20134709001000 2014***4278201100 TOTAL 235971103200 SL Según MSN debería 91-29200 (*) HCTRANSITORIO (* *) SIN TEST DEL SUDOR *** hasta octubre
PKU (+)TIR(+)17-OH Pg (+)GALACTOSA BIOTINI DASA 2009 (sep- dic) (*) (* *) *** TOTAL SL Según MSN debería (*) HCTRANSITORIO (* *) SIN TEST DEL SUDOR *** hasta octubre")
45
Datos propios: Detección de Heterocigotas Screening poblacional para la mutación F 508: 5,65/1000 (1 cada 177). Datos en la Argentina 1:27

Presentaciones similares
>")
 ¿ENFERMEDADES RARAS?>")














 Acido Ribunocleico (ARN) Grandes macromoléculas lineales Función: Transmisión.>")


