Descargar la presentación
La descarga está en progreso. Por favor, espere

1
ENFERMEDADES DEL MUSCULO
BRUNO DE AMBROSI 2008

2
DISTROFIAS MUSCULARES

3
GENERALIDADES Enf degenerativas primarias. Del músculo esquelético.
Base genética.

4
ELEMENTOS COMUNES Debilidad proximal: en mmss y mmii.
Debilidad facial y cervical: sonrisa débil y desagradable. Atrofia y seudo hipertrofia. Mialgia y calambres. EMG miopático. Elevación de la CPK y la aldolasa. Preservación inicial de los ROT. No tnos sensitivos.

5
DISTROFIA DE DUCHENNE

6
GENERALIDADES Destrucción progresiva del músculo por ausencia de distrofina. Prevalencia 3/ Asociada al cr X en forma recesiva por lo que afecta más a varones. 70 % pacientes aumento CPK.

7
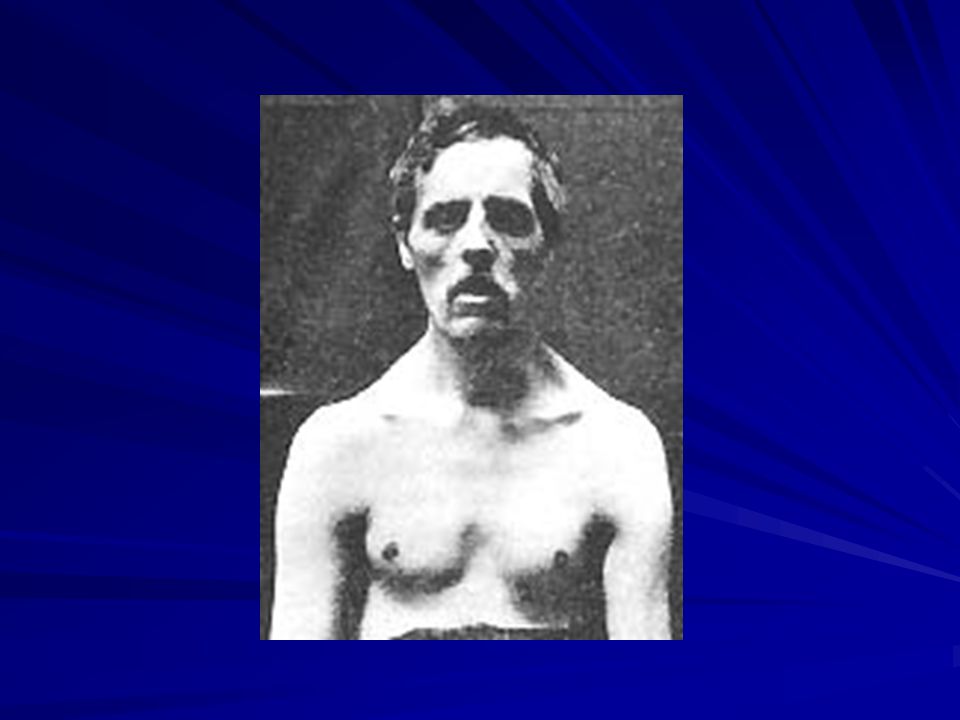
CLINICA Dx 3-6 años. Silla de ruedas 7-14 años
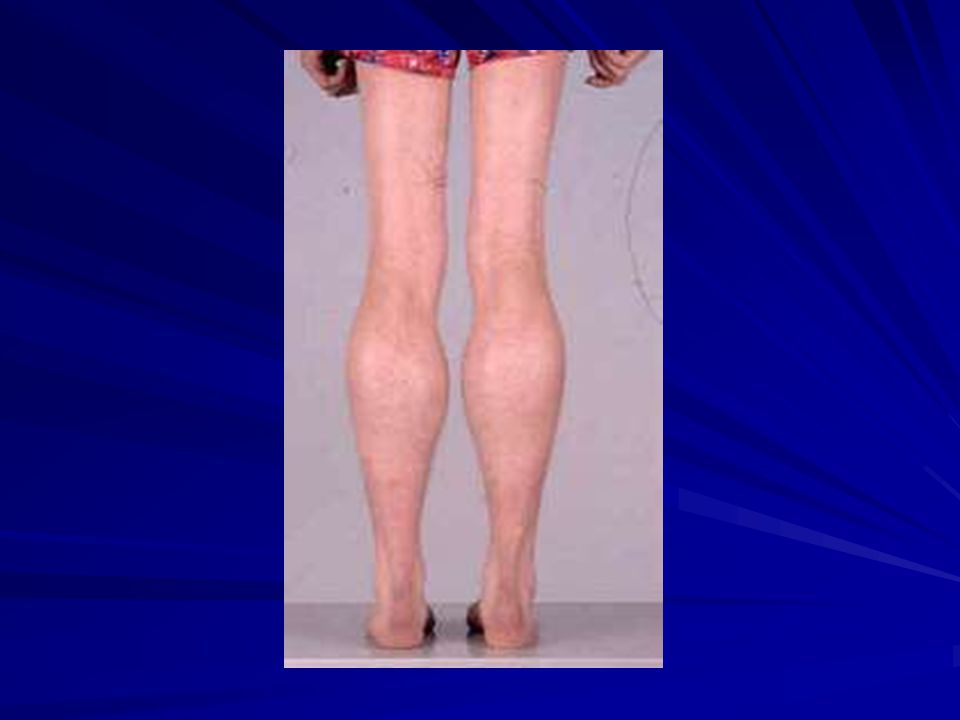
CPK elevada desde el nacimiento. Primer síntoma debilidad. Inicialmente psoas, cuádriceps, glúteos; luego pretibiales, luego mmss, más en proximales (escápula alada). Inicialmente hipertrofia 3-6 a (débiles e hipotónicos) y luego atrofia. Debilidad abdominales y paravertebrales: lordosis y cifosis de sentado.
. Inicialmente hipertrofia 3-6 a (débiles e hipotónicos) y luego atrofia. Debilidad abdominales y paravertebrales: lordosis y cifosis de sentado.")
8
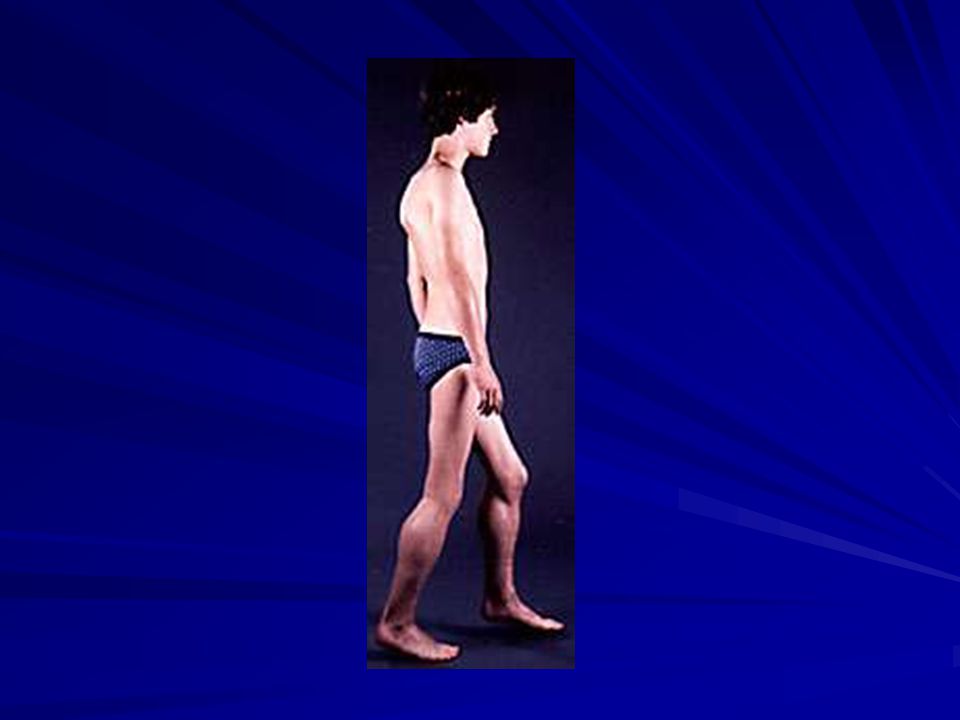
CLINICA Posición: lordosis lumbar, flexión aducción caderas, flexión rodillas, flexión plantar y antebrazos y escoliosis. ROT diminuidos ausentes. Pérdida del músculo cardíaco. Algunos tnos del ritmo cardíaco: taquicardia sinusal, ritmos ectópicos, etc. Frecuente retraso mental leve. Mueren por tnos respiratorios. Afectación músculo GI (seudo obstrucción).
.")
9
From: A Kornberg

11
DIAGNOSTICO CPK. Puede elevarse sin clínica, pico a los 3 años y luego baja. Mioglobina. Biopsia si genético negativo. Distrofina menor al 5% de lo normal. EMG: no específico. Aumento del reclutamiento, PUM miopáticos, aumento de la actividad de inserción. Luego baja el reclutamiento.

12
TRATAMIENTO Corticoides: se desconoce mecanismo pero mejora fuerza y función pulmonar. Prednisona. Rehabilitación. Cirugía. Evitar sedentarismo y obesidad. Ortesis. Ventilación asistida.

13
DISTROFIA DE BECKER

14
GENERALIDADES Comparte el gen con Duchenne, por ende cr X recesivo en hombres. Distrofina presente con alteración de su calidad y/o cantidad. Más benigno que Duchenne.

15
CLINICA Inicio primera década. Patrón similar a Duchenne.
Silla de ruedas a los años. Frecuentemente calambres. Miocardiopatía.

17
DIAGNOSTICO Historia. Genética. Biopsia.

18
TRATAMIENTO Pronóstico similar Duchenne pero más lento. Tratamiento similar.

19
DISTROFIA DE EMERY-DREYFUSS

20
GENERALIDADES Recesiva ligada al cromosoma X. Benigna.
Déficit de emerina.

21
CLINICA Tríada clásica: 1- contractura tobillos, codos y m cervicales posteriores limitantes en el movimiento antes que la debilidad; 2- debilidad y atrofia húmero-perónea y luego en músculos proximales; 3- tnos de la conducción cardíaca (es característica la parálisis auricular). En gral fallecen por bloqueos o insuficiencia cardíaca.
. En gral fallecen por bloqueos o insuficiencia cardíaca.")
22
Rigid spine

23
DIAGNOSTICO CPK moderadamente elevados o normales.
Biopsia cambios distróficos. Confirmación inmunohistoquímica por ausencia de emerina.

24
TRATAMIENTO Fisioterapia, ortesis y cirugía.
Contracturas no son prevenibles. Marcapasos.

25
DISTROFIA FACIOESCAPULOHUMERAL

26
GENERALIDADES Herencia autosómica dominante. Prevalencia 1 en

27
CLINICA Clínica variable en gravedad.
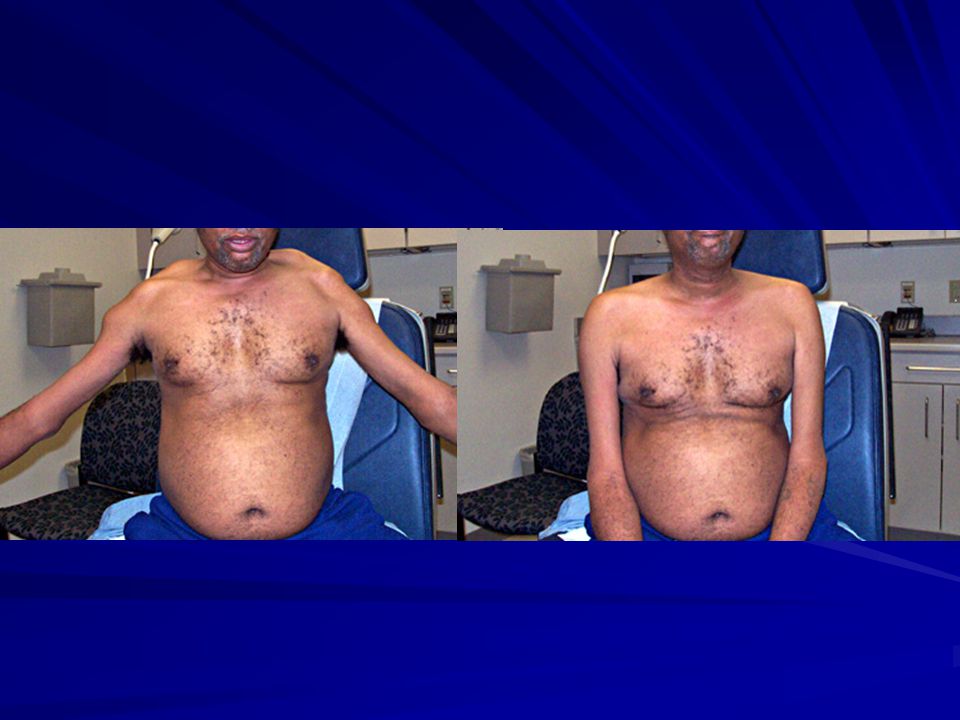
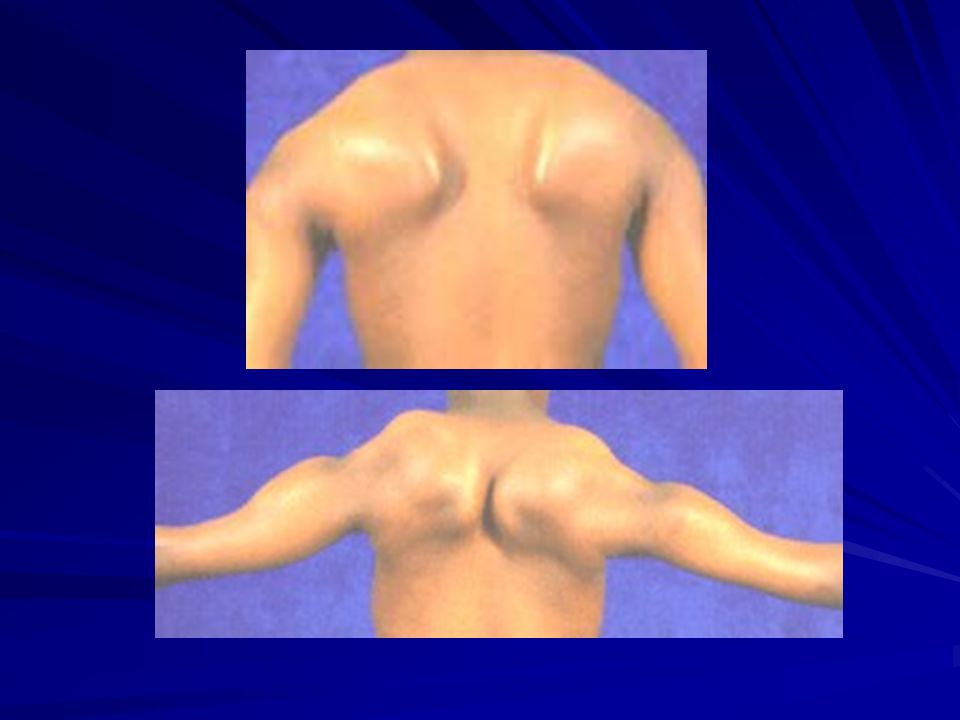
Clásicamente: comienza a los 6-20 años. Debilidad facial (boca de tapir), orbicular labios y ojos. Maseteros, extraoculares y faríngeos no se afectan. Atrofia trapecio y ECM con escápula alada. Atrofia pectorales. Atrofia brazo y no antebrazo (Popeye). Más tardíamente: m de pelvis, cuádriceps y dorsiflexores. En general asimétrico. Raro el compromiso cardíaco.
, orbicular labios y ojos. Maseteros, extraoculares y faríngeos no se afectan. Atrofia trapecio y ECM con escápula alada. Atrofia pectorales. Atrofia brazo y no antebrazo (Popeye). Más tardíamente: m de pelvis, cuádriceps y dorsiflexores. En general asimétrico. Raro el compromiso cardíaco.")
30
DIAGNOSTICO EMG: PUM miopáticos. Biopsia: patrón distrófico.
CPK elevada o normal. Estudio genético.

31
TRATAMIENTO Progresión lenta y supervivencia normal.
Tto de sostén, no específico.

32
Limb girdle dystrophies: Dominant 1A: Myotilin; 5q31; Dysarthria 1B: Lamin A/C; 1q21; + Cardiac 1C: Caveolin-3; 3p25; Child onset 1D: 7q Dilated Cardiomyopathy (?1E): 6q23 1F: 7q32 1G: 4q21 Ankle contractures & High CK Bethlem: Collagen VI; 21q22 & 2q37 Central core: Ryanodine receptor (19q13) Cytoplasmic body: 2q24; 2q21 + Other Distal myopathies MPD2: 5q31; ? Same locus as LGMD1A Emery-Dreifuss: Lamin A/C; 1q21 Facioscapulohumeral: 4q35 Myofibrillar (Desmin storage) Desmin: 2q35; AD or AR αB-crystallin: 11q22 Filamin C: 7q32 LGMD 1A: Myotilin; 5q31 Congenital: SEPN1; 1p36 ZASP myopathy: 10q22 BAG3: 10q25 Other Myotonic (DM1): DMPK; 19q13 Myotonic (DM2): ZNF9; 3q21 Oculopharyngeal: PABP2; 14q11 Skeletal + Myopathy Bone fragility: 9p21 Paget disease: VCP; 9p13 Limb girdle dystrophies: Recessive 2A: Calpain-3 ;15q15 2B: Dysferlin; 2p13.1 2C: γ-Sarcoglycan; 13q12 2D: α-Sarcoglycan; 17q21 2E: β-Sarcoglycan; 4q12 2F: δ-Sarcoglycan; 5q33 2G: Telethonin; 17q11-12 2H: TRIM32; 9q31-q33 2I: FKRP; 19q13.3 2J: Titin; 2q24 2K: POMT1; 9q34 2L: 11p13 2M: Fukutin; 9q31 Merosin (Laminin α2) Absent: 6q2 Reduced: 6q2 Abnormal: LGMD 2I Caveolin-3 mutations Limb girdle dystrophies: X-linked Barth: G4.5 (Tafazzins); Xp28 Becker: Dystrophin; Xp21 Duchenne: Dystrophin; Xp21 Emery-Dreifuss: Emerin; Xq28 Manifesting carriers Dystrophinopathy Myotubularin McLeod Syndrome: XK; Xp21.1; Vacuolar Danon's disease: LAMP-2; Xq24 Excessive Autophagy: Xq28 Mental retardation & Cardiomyopathy
 Absent: 6q2 Reduced: 6q2 Abnormal: LGMD 2I Caveolin-3 mutations Limb girdle dystrophies: X-linked Barth: G4.5 (Tafazzins); Xp28 Becker: Dystrophin; Xp21 Duchenne: Dystrophin; Xp21 Emery-Dreifuss: Emerin; Xq28 Manifesting carriers Dystrophinopathy Myotubularin McLeod Syndrome: XK; Xp21.1; Vacuolar Danon s disease: LAMP-2; Xq24 Excessive Autophagy: Xq28 Mental retardation & Cardiomyopathy.")
33
Other inherited myopathy syndromes Barnes's myopathy Cardiac + Myopathy Cardiomyopathy-associated myopathy Cardiomyopathy (?LGMD1B) Dilated Cardiomyopathy: 6q23 Congenital Myopathies: Late-onset Muscular dystrophies Cytoplasmic body myopathies Distal myopathies Excessive autophagy: Xq28 Familial myasthenia gravis FSH dystrophy: 4q35 Hereditary IBM syndromes IBM1: Dominant IBM2: GNE; 9p12; Recessive IBM3: MyHC-IIa; 17p13; Dominant IBM + Paget disease: 9p13; Dominant Metabolic myopathies Glycogen Lipid Mitochondrial Myopathy + PEO: 17p13; Recessive Myotonic dystrophy Other dystrophies Reducing body Respiratory failure Scapuloperoneal syndromes Skeletal + Myopathy Diaphyseal dysplasia: TGFB1; 19q13 Epiphyseal dysplasia: COL9A3; 20q13 Spheroid body (Myotilin) Tubular aggregates Tubular arrays
 Dilated Cardiomyopathy: 6q23 Congenital Myopathies: Late-onset Muscular dystrophies Cytoplasmic body myopathies Distal myopathies Excessive autophagy: Xq28 Familial myasthenia gravis FSH dystrophy: 4q35 Hereditary IBM syndromes IBM1: Dominant IBM2: GNE; 9p12; Recessive IBM3: MyHC-IIa; 17p13; Dominant IBM + Paget disease: 9p13; Dominant Metabolic myopathies Glycogen Lipid Mitochondrial Myopathy + PEO: 17p13; Recessive Myotonic dystrophy Other dystrophies Reducing body Respiratory failure Scapuloperoneal syndromes Skeletal + Myopathy Diaphyseal dysplasia: TGFB1; 19q13 Epiphyseal dysplasia: COL9A3; 20q13 Spheroid body (Myotilin) Tubular aggregates Tubular arrays")
34
DISTROFIAS DE CINTURAS

35
GENERALIDADES Autosómico recesivo, dominante y esporádicos.
Característica común: debilidad de cinturas sin afectación facial. Inicio infancia tardía y adultez. Inicio escapular o pélvico. Cuanto más tardía más benigna. CPK normal o leve elevación. EMG miopático. No hay afectación cardíaca ni intelectual.

36
DISTROFIA MUSCULAR AUTOSOMICA RECESIVA SEVERA DE LA INFANCIA
Clínica simil Duchenne. Diferencia genética: afecta niños y niñas. Defecto en las glicoproteínas asociadas a distrofina.

37
LIMB GIRDLE DISTROFIAS

38
TIPO PELVIFEMORAL Mayoría esporádica Entre 7 y 49 años.
Inicia en mmii y a los 8 años afecta mmss. Asimetría en un 30 %. CPK por 6.

39
TIPO ESCAPULOHUMERAL Autosómico recesivo.
Inicio primera a cuarta década. Primero a los músculos del hombro, mucho después mmii. Buena sobrevida, lenta evolución. CPK elevada.

40
DISTROFIAS MUSCULARES CONGENITAS

41
GENERALIDADES Desde el nacimiento.
Contracturas de músculos proximales y de tronco. Severidad y evolución variable. CPK elevada. EMG miopático.

42
VARIEDADES Tipo Fukuyama: retraso mental y tnos desarrollo cx cerebral. Tipo Walker-Marburg: anomalías retinianas, lisencefalia. Tipo “músculo-ojo-cerebro”: retina anormal, hidrocefalia, paqui-polimicrogiria, defectos septum pellucidum y cuerpo calloso. Tipo Clásico: muscular exclusivo, 50 % afectación merosina.

43
DISTROFIAS DISTALES

44
Grupo heterogéneo inicio adulto.
Principalmente en extensores sin afectación sensitiva ni pares craneanos. Se dividen en tardías y tempranas, de mmii o mmss, según patrón genético y hallazgos histopatológicos.

45
DISTROFIA MIOTONICA

46
GENERALIDADES Distrofia más frecuente de los adultos.
Autosómica dominante. Más en músculos distales. Fenómeno de anticipación. Prevalencia 5 cada

47
CLINICA En adulto joven.
Debilidad y atrofia de los m distales de mmss. Debilidad y atrofia de los m faciales con ptosis y atrofia de temporales y maseteros. Miotonía principalmente en manos. DBT, impotencia, tnos aprendizaje y cataratas.

48
CLINICA Progresión lenta.
Aspecto característico: calvicie frontal, atrofia maseteros y temporales, de ECM (cuello de cisne), voz nasal, debilidad y atrofia distal. Puede tener cierto retraso mental, apatía e indiferencia. ROT disminuídos. Miotonía es de lengua y músculos distales y no suele ser severa.
, voz nasal, debilidad y atrofia distal. Puede tener cierto retraso mental, apatía e indiferencia. ROT disminuídos. Miotonía es de lengua y músculos distales y no suele ser severa.")
49
CLINICA 90 % opacidad de cristalino. Calcificaciones anormales.
Atrofia testicular. Tnos menstruales. Resistencia insulina. Afectación m liso con afectación de la vía digestiva y de útero. Afectación del sistema de conducción cardíaco.

51
DIAGNOSTICO EMG: descargas miotónicas, patrón miopático.
Biopsia de músculo típica pero no específica. Test genético: cr 19.

52
TRATAMIENTO ECG periódicos en busca prolongación del PR. Marcapasos.
Kinesiología respiratoria. Para la miotonía se utiliza DFH principalmente sino sulfato de quinina o procainamida.

53
MIOTONIAS CONGENITAS

54
THOMSEN Miotonía temprana e hipertrofia.
Miotonía más marcada al despertar o luego del reposo y mejora con la actividad. Más afectados: manos, pies y párpados. Raro debilidad

55
BECKER Más tardía que Thomsen. Más contractura.
Debilidad con la evolución.

56
PARAMIOTONIA CONGENITA
Dominante de alta penetrancia. Miotonía evocada por frío, empeora con actividad (miotonía paradojal) y silencio eléctrico. Debilidad.
 y silencio eléctrico. Debilidad.")
57
MIOPATIAS INFLAMATORIAS

58
De causa autoinmune (polimiositis, dermatomiositis, por cuerpos de inclusión) o por microorganismos (por ej parásitos: toxoplasma, cisiticerco, etc).
 o por microorganismos (por ej parásitos: toxoplasma, cisiticerco, etc).")
59
POLIMIOSITIS Y DERMATOMIOSITIS

60
GENERALIDADES En PM el 20 % tiene enf autoinmune (LES, Sjogren, AR). Vasculitis, Hashimoto, Crohn, LYME, enf celíaca, etc. También HIV y enterovirus. DM se asocia a esclerodermia y enf mixta. PM y DM comparten la inflamación y debilidad muscular pero tiene diferencias inmunológicas y anatomopatológicas.

61
CLINICA PM: precedida en gral por fiebre, con instalación insidiosa.
Se afectan músculos proximales (de las cinturas), los del cuello (ext y flex) y los de la deglución. Poca afectación facial y nunca extraocular. En PM y DM se puede afectar el corazón eléctrica o mecánicamente.
, los del cuello (ext y flex) y los de la deglución. Poca afectación facial y nunca extraocular. En PM y DM se puede afectar el corazón eléctrica o mecánicamente.")
62
CLINICA DM: debilidad subaguda de músculos proximales y flexores del cuello. Afecta más abductores y los extensores. Disfagia en un 30 %. Dolor muscular espontáneo y a la palpación en 50 %. Manifestaciones cutáneas: previo a lo muscular, rash violáceo en parpados y eritematoso en región malar, cuello, hombros u superficies extensoras de mmss e ii. Signo de Gottron: eritema y descamación nudillos. En niños: calcificaciones subcutáneas, vasculitis GI y fibrosis pulmonar.

64
PRONOSTICO DM de 6 a 40 meses. Mortalidad 7 %. Monofásico en gral.
Pronóstico desfavorable: paraneoplásico, vejez, compromiso cardíaco y/o pulmonar, disfagia, inicio agudo.

65
DIAGNOSTICO Clínica. CPK muy elevada, así como la aldolasa, GOT, GPT y LDH. 50 % aumento de VSG, y positividad de ANA y FR. Anti Jo. EMG: miopático inicial, con el tiempo puede ser neurogénico. Biopsia: PM infiltrado inflamatorio, necrosis y fagocitosis, DM atrofia perifascicular e infiltrado perivascular (microangiopatía).
.")
66
TRATAMIENTO Corticoides: prednisona mg/kg/d hasta normalizar la CPK, y disminución de un 20 % por mes o bien disminución a días alternos. En los severos puede usarse metilprednisolona EV por 5 días. Se asocian con pérdida de fibras tipo 2. Si no responden a corticoides: azatioprina, metotrexato, ciclofosfamida, ciclosporina, clorambucil. Plasmaféresis sin clara demostración. Inmunoglobulina.

67
MIOSITIS POR CUERPO DE INCLUSION

68
GENERALIDADES Miopatía más frecuente en hombres mayores de 50 años.
Esporádica.

69
CLINICA Debilidad de instalación subaguda y progresión lenta.
Asimétrica, tanto proximal como distal. Más afectados: cuádriceps, flexores muñeca y dedos, dorsiflexores pies. 40% disfagia y músculos faciales. 30% neuropatía. No m extraoculares.

70
DIAGNOSTICO CPK por 10. EMG : patrón neuropático y miogénico.
Anatomía patológica.

71
TRATAMIENTO Prednisona altas dosis: leve beneficio.
Algunos trabajos con IVIg. Azatioprina.

Presentaciones similares