Descargar la presentación
La descarga está en progreso. Por favor, espere

1
Modelamiento de Proteinas
Dinámica molecular y análisis de la energía libre de Interacciones inhibidor cathepsin D: Penetración en diseño de Structure-Based Ligando. Shuanghong Huo, Modelamiento de Proteinas

2
INTRODUCCION - Se realizo Estudios en un grupo de I. CAT D de E°L. Por MD. Método de Solvente Continuo. (MM-PBSA). 07 I. sintetizados por quimica combinatorial, con e/p = 1Kcal/mol y cc. =0.98 - El docking tanto en Rayos – X, y MD son concordantes, para un Péptido inhibidor unido a Cat D. geometricamente. - Sin embargo, la función DOCK scoring basado en Fuerzas Vdw y Electrostáticas Utilizan una Cte Dieléctrica dependiente en distancias reproduce una pobre tendencia experimental de afinidad de unión a estos Is. Finalmente el uso de análisis PROFEC, permitió identificar dos sustituciones posibles para mejorar la unión a uno de los mejores inhibidores de Cat D

3
INTRODUCCION Catepsina D : Proteína Aspartica lisosomal, puede cortar precursores proteínicos β-amiloideos para liberar peptidos alzeimer. Implicado en cáncer: de mamas y ovárico. Diseño de estructuras junto con química combinatorial ha sido exitosamente usado para el diseño de inhibidores no peptidicos de Cat D. Tomando como modelo la estructura obtenida con R- X, la catepsina humana con su I. pepstatina, se diseño una estructura con el algoritmo COMBIBUILD, a fin de buscar una nueva conformación optima estructural. Nuevos métodos han sido desarrollados para calcular la E°L. en forma rápida y practica. MD, MM-PBSA, LIE (Energía Lineal de Interacción), Empíricos como LUDI. LIE, método semiempirico, propuesto por Aqvist. (calcula el Peso de Interacciones Electrostáticas y vdW). Interacciones entre el ligando y el receptor.
, Empíricos como LUDI. LIE, método semiempirico, propuesto por Aqvist. (calcula el Peso de Interacciones Electrostáticas y vdW). Interacciones entre el ligando y el receptor.")
4
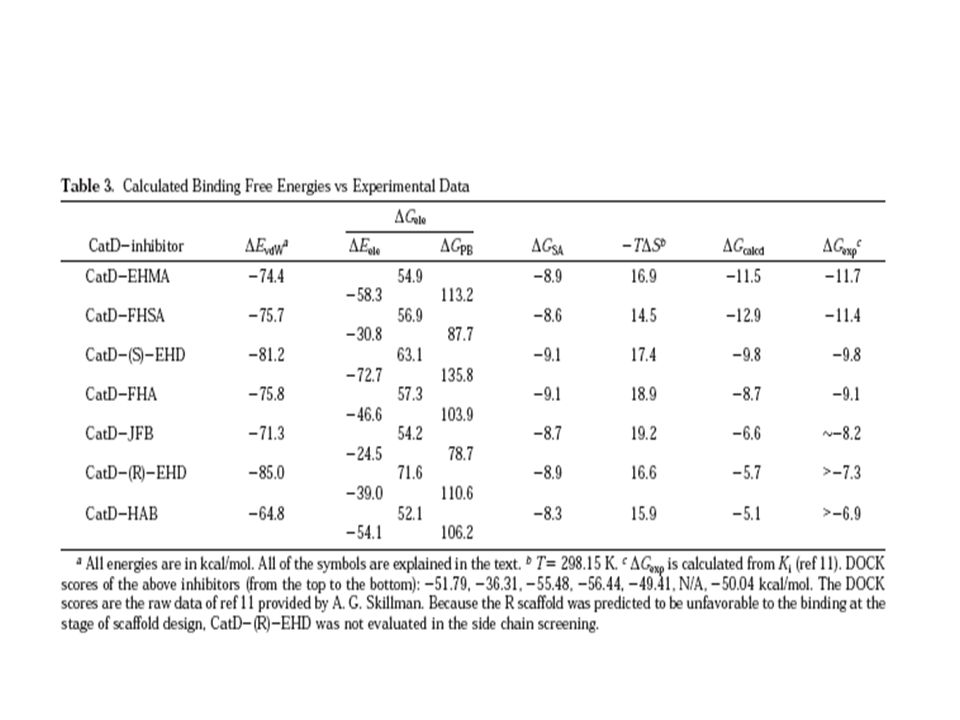
Objetivo 1.- Si el método MM-PBSA proporciona una evaluación cuantitativa de la unión de un grupo de compuestos de CatD con parámetros no adjuntos de los factores de peso de las interacciones E° y vdW Exploraron modificaciones de estos compuestos que pueden dar alta afinidad de union a ligandos. Calculo de la unión del diseño de inhibidores de CatD usando el método MM-PBSA. Que combina modelos de solvatacion internos y externos, por (PB).
.")
5
Modelo mecánico molecular de los inhibidores
Parm94 (AMBER) no incluye cargas Optimización geométrica con AM1 RHF/6-31G cálculo de punto individual con Gaussian 98 (potenciales electrostáticos). Método RESP para el acomodo de las cargas de cada inhibidor Parm99 (parametros de enlace, angulo, angulo de torsion y vdW)
 no incluye cargas. Optimización geométrica con AM1. RHF/6-31G cálculo de punto individual con Gaussian 98 (potenciales electrostáticos). Método RESP para el acomodo de las cargas de cada inhibidor. Parm99 (parametros de enlace, angulo, angulo de torsion y vdW)")
8
y =0.92 kcal/mol b= 0.00542 kcal/mol A°2

9
Conformación inicial del complejo CatD-EHMA
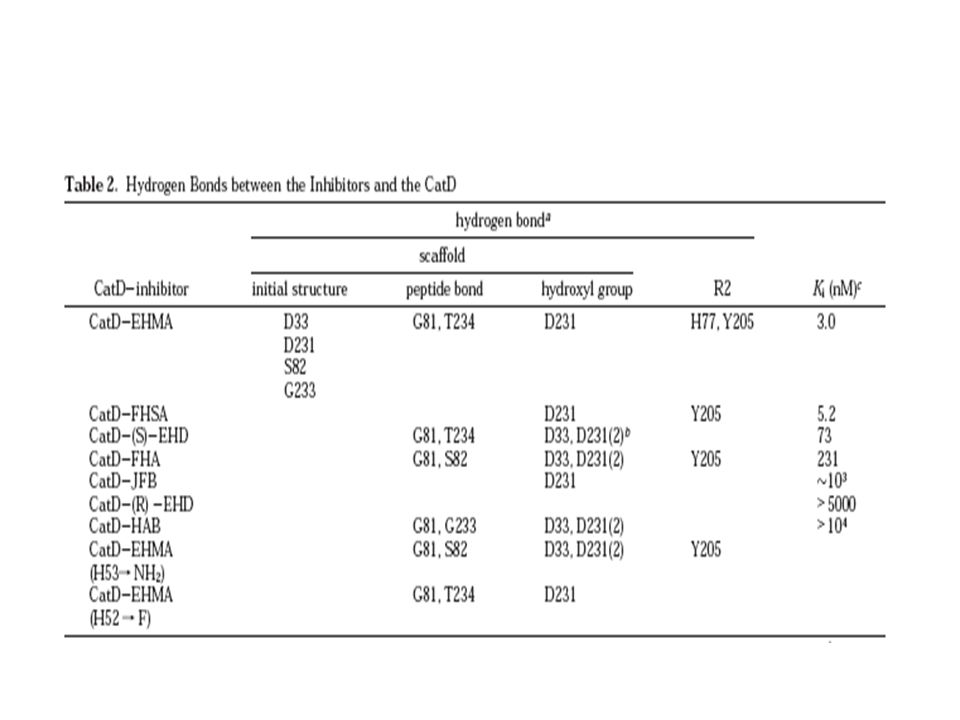
D33 (cadena A) , D231 (cadena B)
 , D231 (cadena B)")
10
EHD(R) epímero
 epímero")
15
R1: no modificaciones R3: oxígeno de unión de R3, podría ser sustituido por un átomo positivamente cargado, pero…. La conformación dicloro-fenilo está restringida por el oxigeno de unión. Debido al gran cambio conformacional que causaría reemplazar el oxígeno de unión, esto se rechaza

16
R4 Reemplazo de H con grupos grandes para R4 y grupos cargados (+) al fenilo Ellman: inhibidor con sustituyentes grandes tolerados por rearreglos de la proteína que PROFEC no detecta R1 R2 R3 R4

17
D75: potencial aceptor de H Simulación MD
H53 NH2 (+) H52 F (-) D75: potencial aceptor de H Simulación MD R1 R2 R3 R4
 H52 F (-) D75: potencial aceptor de H. Simulación MD. R1. R2. R3. R4.")
18
H53: Desolvatación: penalty incremetada
La conformación del ligando cambia La red de enlaces de hidrógenos cambia Tabla2 El en electrostática se contrarresta con el alto costo de desolvatación: 12,2 kcal/mol H52: en interacciones vdW, pero las interacciones electrostáticas son menos favorables 9,6, el costo de desolvatación es un poco menor

19
Conclusiones Se ha estudiado la energía de unión
Los resultados de las simulaciones MD coincidieron con las estructuras cristalizadas

Presentaciones similares













>")

>")

 Sintetizado en vivo por la enzima NO-sintetasa>")





