Descargar la presentación
La descarga está en progreso. Por favor, espere

1
QUIMICA FISICA BIOLOGIA TEMA 4-EQUILIBRIO HETEROGENEO
QUIMICA FISICA I 2013 TEMA 4-EQUILIBRIO HETEROGENEO

2
Equilibrio de fases en sistemas simples. La condición de equilibrio
Para un sistema en equilibrio, el potencial químico (μ) de cada constituyente debe ser el mismo en cualquier parte del sistema. Si hay varias fases presentes, μ de cada sustancia debe tener el mismo valor en cada fase en la cual se presenta la sustancia Para un sistema de un componente, μ=G/n, así tenemos: Donde son la entropía molar y el volumen molar: Entonces Las derivadas de estas ecuaciones son las pendientes de las curvas μ contra T y μ contra p, respectivamente. (A)
 de cada constituyente debe ser el mismo en cualquier parte del sistema. Si hay varias fases presentes, μ de cada sustancia debe tener el mismo valor en cada fase en la cual se presenta la sustancia. Para un sistema de un componente, μ=G/n, así tenemos: Donde son la entropía molar y el volumen molar: Entonces. Las derivadas de estas ecuaciones son las pendientes de las curvas μ contra T y μ contra p, respectivamente. (A)")
3
Estabilidad de las fases de una sustancia pura
Según la tercera ley de la termodinámica, la entropía de una sustancia es siempre positiva. Este hecho, combinado con la ecuación (1), muestra que es siempre negativa. En consecuencia, la grafica de μ contra T a presión constante es una curva con una pendiente negativa. Para las tres fases de una sustancia simple tenemos: (3) A cualquier temperatura, La entropía del sólido es pequeña, de modo que la curva μ contra T para el sólido, curva S en la figura 1, tiene una pendiente ligeramente negativa. La curva μ contra T para el líquido tiene una pendiente un poco más negativa que la del sólido, curva L. La entropía del gas es mucho mayor que la del líquido, de aquí que la pendiente de la curva G tiene un valor negativo mayor. Las condiciones termodinámicas para el equilibrio entre fases a presión constante se deducen fácilmente de la figura 1
, muestra que es siempre negativa. En consecuencia, la grafica de μ contra T a presión constante es una curva con una pendiente negativa. Para las tres fases de una sustancia simple tenemos: (3) A cualquier temperatura, . La entropía del sólido es pequeña, de modo que la curva μ contra T para el sólido, curva S en la figura 1, tiene una pendiente ligeramente negativa. La curva μ contra T para el líquido tiene una pendiente un poco más negativa que la del sólido, curva L. La entropía del gas es mucho mayor que la del líquido, de aquí que la pendiente de la curva G tiene un valor negativo mayor. Las condiciones termodinámicas para el equilibrio entre fases a presión constante se deducen fácilmente de la figura 1.")
4
Figura 1.μ contra T a presión constante

5
El estado sólido y el líquido coexisten en equilibrio cuando μsólido= μlíquido ; esto es, en los puntos de intersección de las curvas S y L. La temperatura es Tm, o sea la de fusión. Análogamente, el estado líquido y el gaseoso coexisten en equilibrio a la temperatura Tb, el punto de intersección de las curvas L y G, al cual μgas= μlíquido El eje de T esta dividido en tres zonas o intervalos. Por debajo Tm el sólido tiene el potencial químico más bajo. Entre Tm y Tb el líquido tiene el mínimo potencial químico. Por encima de Tb el gas tiene el potencial químico más bajo. La fase con el potencial químico más bajo es la fase estable. Si la fase líquida estuviese presente en un sistema a una temperatura menor de Tm, Figura 2, el potencial químico del líquido tendría el valor μa, mientras que el sólido tendría el valor μb. Así, el líquido podría congelarse espontáneamente a esta temperatura, debido a que el congelamiento disminuiría su potencial químico. A una temperatura mayor que Tm la situación es inversa; el μsólido es mayor que la del líquido y el sólido se funde espontáneamente, disminuyendo así el potencial químico del sistema. En Tm, los potenciales químicos del sólido y del líquido son iguales, es decir ninguna de las fases predomina, coexisten en equilibrio.

6
Dependencia de las curvas de μ contra T, con la presión
Que acontece si se cambia la presión?. Se responde aplicando la ecuación (2) en la forma dμ=Vdp. Si disminuye la presión, dp es negativa, V es positivo, por lo tanto dμ es negativo y el potencial químico disminuye proporcionalmente al volumen de la fase. Como los V molares del líquido y el sólido son pequeños el valor de μ disminuye muy poco, para el sólido de a hasta a’, para el líquido de b hasta b’. Figura 3 a. El volumen del gas es 1000 veces mayor que el del sólido o el del líquido, por eso μ del gas disminuye considerablemente de c hasta c’. Las curvas a presiones menores se indican en la Figura 3 b, como líneas discontinuas paralelas a las originales. En consecuencia vemos que se han desplazado ambas T de equilibrio, que el desplazamiento de la Tm es menor que la de Tb, que el intervalo de estabilidad de un líquido disminuye apreciablemente, que si disminuye mucho la presión la Tb puede caer por debajo de la Tm, entonces no hay temperatura a la cual el líquido sea estable, el sólido sublima, a esa temperatura Ts el sólido y el vapor coexisten en equilibrio. Figura 4
 en la forma dμ=Vdp. Si disminuye la presión, dp es negativa, V es positivo, por lo tanto dμ es negativo y el potencial químico disminuye proporcionalmente al volumen de la fase. Como los V molares del líquido y el sólido son pequeños el valor de μ disminuye muy poco, para el sólido de a hasta a’, para el líquido de b hasta b’. Figura 3 a. El volumen del gas es 1000 veces mayor que el del sólido o el del líquido, por eso μ del gas disminuye considerablemente de c hasta c’. Las curvas a presiones menores se indican en la Figura 3 b, como líneas discontinuas paralelas a las originales. En consecuencia vemos que se han desplazado ambas T de equilibrio, que el desplazamiento de la Tm es menor que la de Tb, que el intervalo de estabilidad de un líquido disminuye apreciablemente, que si disminuye mucho la presión la Tb puede caer por debajo de la Tm, entonces no hay temperatura a la cual el líquido sea estable, el sólido sublima, a esa temperatura Ts el sólido y el vapor coexisten en equilibrio. Figura 4.")
7
Figura 3. Efecto de la presión en las temperaturas de fusión y ebullición. Las líneas continuas indican altas presiones, las discontinuas bajas presiones

8
Figura 4. μ contra T para una sustancia que sublima

9
La ecuación de Clapeyron
La condición de equilibrio entre dos fases, α y β, de una sustancia pura es μα(T,p) = μβ(T,p) (4) Si conociésemos las formas analíticas de las funciones μα y μβ, sería posible resolver la ecuación (4) para: T=f (p) (5) o p=g (T) (6) La ecuación (5) expresa el hecho ilustrado en la figura 3b, de que la T de equilibrio depende de p. Si las funciones μα y μβ no se conocen detalladamente, es posible, no obstante obtener un valor para la derivada de la temperatura con respecto a la presión. Consideremos el equilibrio entre las fases α y β bajo una presión p, la temperatura de equilibrio es T, se cumple la ecuación (4)
 = μβ(T,p) (4) Si conociésemos las formas analíticas de las funciones μα y μβ, sería posible resolver la ecuación (4) para: T=f (p) (5) o p=g (T) (6) La ecuación (5) expresa el hecho ilustrado en la figura 3b, de que la T de equilibrio depende de p. Si las funciones μα y μβ no se conocen detalladamente, es posible, no obstante obtener un valor para la derivada de la temperatura con respecto a la presión. Consideremos el equilibrio entre las fases α y β bajo una presión p, la temperatura de equilibrio es T, se cumple la ecuación (4)")
10
μα(T,p) + dμα= μβ(T,p)+dμβ (7)
Si la presión se cambia hasta el valor p+dp, la temperatura de equilibrio será T+dT y el valor de μ cambiará a μ+dμ. Por lo tanto, a T+dT, p+dp la condición de equilibrio es: μα(T,p) + dμα= μβ(T,p)+dμβ (7) Sustrayendo la ecuación (4) de la (7) obtenemos que: dμα= dμβ (8) Ahora expresamos dμ en términos de dp y dT, aplicando la ecuación fundamental (A) Empleando las ecuaciones (9) en la ecuación (8), obtenemos: Reordenando tenemos:
 + dμα= μβ(T,p)+dμβ (7) Sustrayendo la ecuación (4) de la (7) obtenemos que: dμα= dμβ (8) Ahora expresamos dμ en términos de dp y dT, aplicando la ecuación fundamental (A) Empleando las ecuaciones (9) en la ecuación (8), obtenemos: Reordenando tenemos:")
11
Esto si la transformación se expresa de αβ, entonces la ecuación (10) se transforma en:
Cualquiera de las ecuaciones anteriores es conocida como ecuación de Clapeyron. Expresa la dependencia cuantitativa de la temperatura de equilibrio con la presión ecuación (12) o la variación de la presión de equilibrio con la temperatura ecuación (13).
 o la variación de la presión de equilibrio con la temperatura ecuación (13).")
12
dP/dT = DHtran/T DVtran
En un cambio de fase reversible (de equilibrio), La ecuación de Clapeyron-Clausius pasa a ser dP/dT = DHtran/T DVtran En una transición líquido- vapor tanto H como V son positivos, por lo tanto dP/dT es positivo, la línea líquido – vapor de un diagrama de fase P-T para un sistema de un componente tiene pendiente positiva. Lo mismo se cumple para la línea sólido-vapor. En una transición líquido-sólido, H es siempre prácticamente positivo; V suele ser positivo, pero en pocos casos es negativo, por ejemplo agua, Ga y Bi. Debido a la disminución de volumen que se produce al fundirse el hielo, la línea de equilibrio sólido-líquido se inclina hacia la izquierda en el diagrama P-T del agua.
, La ecuación de Clapeyron-Clausius pasa a ser. dP/dT = DHtran/T DVtran. En una transición líquido- vapor tanto H como V son positivos, por lo tanto dP/dT es positivo, la línea líquido – vapor de un diagrama de fase P-T para un sistema de un componente tiene pendiente positiva. Lo mismo se cumple para la línea sólido-vapor. En una transición líquido-sólido, H es siempre prácticamente positivo; V suele ser positivo, pero en pocos casos es negativo, por ejemplo agua, Ga y Bi. Debido a la disminución de volumen que se produce al fundirse el hielo, la línea de equilibrio sólido-líquido se inclina hacia la izquierda en el diagrama P-T del agua.")
13
Integración de la ecuación de Clapeyron
EQUILIBRIO LÍQUIDO-VAPOR Y SÓLIDO-VAPOR En el equilibrio V-L ó V-S, el Vm,vapor >> Vm,líq ó Vm,sól a menos que T esté cercana a la temperatura crítica, en cuyo caso las densidades del líquido y el vapor son parecidas. Por eso cuando una de las fases es un gas, Vm= Vm,vapor -Vm,líq ó sól Vm,vapor . Si este se comporta como ideal Esta dos aproximaciones nos conducen a Reemplazando en la ec. de Clapeyron-Clausius Equilibrio S-G o L-G lejos de Tc

14
EQUILIBRIO LÍQUIDO-SÓLIDO
La ec. Anterior no tiene validez en una transición sólido-líquido. Durante la fusión la ec. De Clapeyron-Clausius es Si separo variables e integro: Las magnitudes y cambian a lo largo de la curva de equilibrio S-L, como consecuencia de los cambios que sufren Tfus y Pfus en esa curva. La elevada inclinación de la curva de fusión en el diagrama P-T, implica que debe ocurrir un gran cambio en la presión para ocasionar pequeños cambios en la T. Puedo así considerar que Hfus y Vfus permanecen ctes. Equilibrio sólido - líquido

15
Diagrama de fases del agua
El punto triple conlleva la definición de la temperatura termodinámica (eficiencia 1 del ciclo de Carnot y intervalos de 1 K a 1/ de la temperatura del punto triple del agua. El hielo es menos denso que el agua (pendiente P/T < 0) El punto de equilibrio de fases a 121 °C y 2 atm es la condición de trabajo de las autoclaves.
 El punto de equilibrio de fases a 121 °C y 2 atm es la condición de trabajo de las autoclaves.")
16
Diagrama de fases del CO2
No hay CO2 líquido a presión y temperatura ambiente. Pendiente P/T > 0, como casi todas las sustancias. El CO2 supercrítico (Tc = 32 °C y Pc = 73 atm) se usa en cromatografía de fluido supercrítico y para extraer la cafeína del café. Casos interesantes: los extinguidores a CO2 y el planeta Marte.
 se usa en cromatografía de fluido supercrítico y para extraer la cafeína del café. Casos interesantes: los extinguidores a CO2 y el planeta Marte.")
17
REGLA DE LAS FASES Una fase es una porción homogénea de un sistema. Un sistema puede tener un número arbitrario de fases sólidas y líquidas, pero una sola fase gaseosa. Para describir el estado de equilibrio de un sistema con varias fases y diversas especies químicas, podemos especificar el número de moles de cada especie en cada una de las fases, además de la T y la P. Si no existen paredes rígidas o diatérmicas separando las fases la T y P son las mismas para todas las fases en equilibrio. No especificaremos el número de moles. La masa o tamaño de cada fase no afecta la posición del equilibrio de fases, ya que viene determinada por la igualdad de potenciales químicos. Nos ocuparemos de las fracciones molares Grado de libertad de un sistema en equilibrio como el número de variables intensivas independientes necesarias para especificar su estado intensivo. .

18
El .estado intensivo de equilibrio se describe especificando las variables intensivas P, T y fracciones molares en cada una de las fases. No todas estas variables son independientes entre si. Para empezar hacemos 2 suposiciones: No ocurre ninguna reacción química Todas las especies químicas están presentes en todas las fases Sea C el número de especies químicas diferentes presentes en el sistema y sea F el número de fases presentes. Según la suposición 2 existen C especies químicas en cada fase, por lo tanto tenemos FC fracciones molares. Añadiendo P y T, tenemos: FC+2 variables intensivas para describir el estado intensivo del sistema en equilibrio. Pero no todas la FC+2 variables son independientes, existen relaciones entre ellas

19
(A) Existe una relación como la anterior para cada fase, por lo que tendremos en total F ecuaciones como la anterior. Podemos así resolver estas ecuaciones para eliminando así F variables intensivas En segundo lugar tenemos las condiciones de equilibrio, los potenciales químicos han de cumplir las condiciones de equilibrio de fase siguientes: (B) Como hay F fases, incluye F-1 signos de igualdad, por lo tanto F-1 ecuaciones independientes. Como existen C especies químicas diferentes, hay un total de C(F-1) signos de igualdad en el conjunto de ecuaciones, por lo tanto hay C(F-1) relaciones independientes entre los potenciales químicos.
 Como hay F fases, incluye F-1 signos de igualdad, por lo tanto F-1 ecuaciones independientes. Como existen C especies químicas diferentes, hay un total de C(F-1) signos de igualdad en el conjunto de ecuaciones, por lo tanto hay C(F-1) relaciones independientes entre los potenciales químicos.")
20
Empezábamos con FC+2 variables intensivas, eliminamos F de ellas de acuerdo a (A) y C(F-1) de acuerdo a (B), entonces el número de variables intensivas independientes es: Regla de las fases sin reacciones REGLA DE LAS FASES EN SISTEMAS REACCIONANTE: Si prescindimos de la suposición 1 y admitimos la posibilidad de que se produzcan reacciones químicas. Por cada reacción química independiente aparece la condición Cada reacción química independiente proporciona una relación entre potenciales químicos y al igual que las (B) cada una de las relaciones se puede utilizar para eliminar una variable de las formadas por P, T y las fracciones molares. Si el número de reacciones independientes es r:
 cada una de las relaciones se puede utilizar para eliminar una variable de las formadas por P, T y las fracciones molares. Si el número de reacciones independientes es r:")
21
Pueden existir otras restricciones en el número de variables intensivas independientes de un sistema, como ser condiciones estequiométricas o de electroneutralidad y la denominamos a

22
Regla de las fases = C – F + 2
Para caracterizar completamente un sistema termodinámico es necesario conocer el valor de un número de variables intensivas independientes = C – F + 2 Sustancia pura: C = 1 F = L = = T y P F = L = = T (o P) F = L = = 0
 F = 3 L = = 0.")
23
Cuando en el sistema ocurren r reacciones químicas, el número de variables independientes se reducen
= C – F + 2-r Si además existen relaciones estequiométricas o de conservación de la electroneutralidad, el número de variables intensivas independientes es menor = C – F + 2-r-a Mezcla gaseosa : N2, H2 y NH3: C = 3 F = = = T , P, X1 y X2 Mezcla gaseosa : N2, H2 y NH3 con catalizador C = F = 1 r = NH3 N2 +3 H2 L = 3 – – 1 = T, P, X1 (KP)
")
24
DIAGRAMA DE UN COMPONENTE
Un diagrama de fase muestra las condiciones de P y T donde las fases son estables y los puntos donde hay transición de fase. C=1 y =2(P y T)
")
25
En este diagrama se ve el efecto de la temperatura y de la presión en una sustancia en un recipiente cerrado. Cada punto en este diagrama representa una combinación posible de la temperatura y de la presión para el sistema. El diagrama se divide en tres áreas, que representan los estados sólidos, líquidos, y gaseosos de la sustancia. La regla de las fases para un sistema de un componente es: =3-F; Si F=1, entonces =2; si F=2, =1, si F=3, =0. el valor máximo de =2. Para un sistema de un componente, su estado intensivo viene descripto al especificar un máximo de dos variables intensivas. Podemos representar cualquier estado intensivo de un sistema de un componente mediante un punto en un diagrama bidimensional de P frente a T, en el que cada punto corresponde a valores definidos de P y T. Este diagrama se denomina diagrama de fases.

26
Las regiones de una fase son las superficies abiertas, en ellas F=1 y existen 2 grados de libertad, ya que para describir el estado intensivo es necesario definir tanto P como T. A lo largo de las líneas (con excepción del punto B) encontramos dos fases en equilibrio, por lo tanto =1 a lo largo de las líneas. Por ejemplo, con liquido y vapor en equilibrio, podemos modificar la T en cualquier punto a lo largo de la línea BC, pero una vez elegido el valor de T, entonces P, la presión de vapor (de equilibrio) de la sustancia pura liquida a la temperatura T, también queda fijada. El punto de ebullición de un liquido a una presión dada P a la temperatura a la cual la presión de vapor de equilibrio es igual a P. El punto de ebullición normal es la temperatura a la cual la presión de vapor del liquido es igual a 1atm. La línea BC proporciona el punto de ebullición de la sustancia pura en función de la presión.
 encontramos dos fases en equilibrio, por lo tanto =1 a lo largo de las líneas. Por ejemplo, con liquido y vapor en equilibrio, podemos modificar la T en cualquier punto a lo largo de la línea BC, pero una vez elegido el valor de T, entonces P, la presión de vapor (de equilibrio) de la sustancia pura liquida a la temperatura T, también queda fijada. El punto de ebullición de un liquido a una presión dada P a la temperatura a la cual la presión de vapor de equilibrio es igual a P. El punto de ebullición normal es la temperatura a la cual la presión de vapor del liquido es igual a 1atm. La línea BC proporciona el punto de ebullición de la sustancia pura en función de la presión.")
27
Si se considera a T como la variable independiente, la Línea BC proporciona la presion de vapor de la sustancia pura en funcion de la temperatura. El punto C de esta curva es el punto crítico, punto en el cual no se puede hacer la diferencia entre estado líquido y estado gaseoso(uno habla de fluido supercrítico) El punto B es el punto triple, punto donde el solido, el liquido y el vapor se encuentran en equilibrio mutuo y =0. Como no existen grados de libertad, el punto triple aparece a valores definidos de P yT. La línea BD es la curva de equilibrio líquido-sólido y se llama curva de fusión y es la que da los valores de la presión de vapor del sólido en función de la temperatura o los puntos de fusión en función de la presión.
 El punto B es el punto triple, punto donde el solido, el liquido y el vapor se encuentran en equilibrio mutuo y =0. Como no existen grados de libertad, el punto triple aparece a valores definidos de P yT. La línea BD es la curva de equilibrio líquido-sólido y se llama curva de fusión y es la que da los valores de la presión de vapor del sólido en función de la temperatura o los puntos de fusión en función de la presión.")
28
La línea de BD es casi vertical porque el punto de fusión de un sólido no es muy sensible a los cambios en la presión. Para la mayoría de los compuestos, esta línea presenta una pendiente positiva pequeña, según lo muestra la figura. Debe tenerse en mente que la pendiente de esta línea es levemente negativa para el caso particular del agua. Consecuentemente, el agua se puede derretir a temperaturas cercanas a su punto de congelación cuando está sujeta a la presión. La facilidad con la cual los patinadores del hielo se deslizan a través de una lago congelado se puede explicar por el hecho de que la presión ejercida por sus patines derrite una porción pequeña del hielo y se forma una superficie líquida entre el hielo y sus patines..

29
A lo largo de la Línea AB existe equilibrio entre el solido y el vapor, la línea AB es la curva de presión de vapor del solido.

30
EQUILIBRIO DE FASES EN SISTEMAS MULTICOMPONENTES
Los equilibrios entre fases pueden corresponder a los más variados tipos de sistemas heterogéneos: un líquido en equilibrio con su vapor, una solución saturada en equilibrio con el soluto en exceso, dos líquidos parcialmente solubles el uno en el otro, dos sólidos totalmente solubles en equilibrio con su fase fundida, dos sólidos parcialmente solubles en equilibrio con un compuesto formado entre ellos, etc. El objetivo es describir completamente el sistema. El comportamiento de estos sistemas en equilibrio se estudia por medio de gráficos que se conocen como diagramas de fase : se obtienen graficando en función de variables como presión, temperatura y composición y el sistema en equilibrio queda definido para cada punto (los gráficos de cambio de estado físico ó de presión de vapor de una solución de dos líquidos son ejemplos de diagramas de fases).
.")
31
La mayoría de los diagramas de fase han sido construidos según condiciones de equilibrio (condiciones de enfriamiento lento), siendo utilizadas por ingenieros y científicos para entender y predecir muchos aspectos del comportamiento de materiales. A partir de los diagramas de fase se puede obtener información como: 1.- Conocer que fases están presentes a diferentes composiciones y temperaturas bajo condiciones de enfriamiento lento( equilibrio). 2.- Averiguar la solubilidad, en el estado sólido y en el equilibrio, de un elemento ( o compuesto) en otro. 3.- Determinar la temperatura en la cual una aleación enfriada bajo condiciones de equilibrio comienza a solidificar y la temperatura a la cual ocurre la solidificación. 4.- Conocer la temperatura a la cual comienzan a fundirse diferentes fases.
. 2.- Averiguar la solubilidad, en el estado sólido y en el equilibrio, de un elemento ( o compuesto) en otro. 3.- Determinar la temperatura en la cual una aleación enfriada bajo condiciones de equilibrio comienza a solidificar y la temperatura a la cual ocurre la solidificación. 4.- Conocer la temperatura a la cual comienzan a fundirse diferentes fases.")
32
Los equilibrios de fase y sus respectivos diagramas de fase en sistemas multicomponentes tienen aplicaciones importantes en química, geología y ciencia de los materiales. La ciencia de materiales estudia la estructura, propiedades y aplicaciones de los materiales científicos y tecnológicos

33
EQUILIBRIO LIQUIDO-LIQUIDO EN SISTEMA DE DOS COMPONENTES
Al existir dos componentes en el sistema en consideración la regla de las fases queda: = C – F + 2=4-F Luego, para representar gráficamente el campo de estabilidad de una región homogénea (monofásica) se requieren 3 variables, lo que hace necesario el sistema en un diagrama tridimensional.
 se requieren 3 variables, lo que hace necesario el sistema en un diagrama tridimensional.")
34
Por lo tanto, 1 fase: bivariante ( =2)
2 fases: univariante ( =1) 3 fases: invariante ( =0) Por conveniencia se suelen mantener P o T constantes y se representa gráficamente un sistema de fases bidimensionales, que es un corte transversal de la representación tridimensional. Siempre que se agitan cantidades arbitrarias del alcohol y agua en un embudo de separación a temperatura ambiente, se obtiene un sistema con una sola fase líquida. El agua y el etanol son solubles entre si en cualquier proporción, por lo que se dice que son totalmente miscibles
 3 fases: invariante ( =0) Por conveniencia se suelen mantener P o T constantes y se representa gráficamente un sistema de fases bidimensionales, que es un corte transversal de la representación tridimensional. Siempre que se agitan cantidades arbitrarias del alcohol y agua en un embudo de separación a temperatura ambiente, se obtiene un sistema con una sola fase líquida. El agua y el etanol son solubles entre si en cualquier proporción, por lo que se dice que son totalmente miscibles.")
35
Cuando se agitan cantidades similares de CH3(CH2)3OH y agua a temperatura ambiente, se obtiene un sistema formado por dos fases líquidas: una de ellas es agua con una pequeña cantidad de CH3(CH2)3OH disuelto, y la otra es CH3(CH2)3OH con una pequeña cantidad de agua disuelta. Estos dos líquidos son parcialmente miscibles, lo que significa que cada uno se disuelve en el otro hasta alcanzar un límite máximo. Cuando se mantiene la P constante (1atm.), la forma más común del diagrama de fases líquido-líquido de T frente a xA para dos líquidos parcialmente miscibles A y B, es como el que se ve en la siguiente figura:
, la forma más común del diagrama de fases líquido-líquido de T frente a xA para dos líquidos parcialmente miscibles A y B, es como el que se ve en la siguiente figura:")
36
fig: diagrama de fases líquido-líquido de temperatura frente a composición para dos líquidos parcialmente miscibles.

37
Supongamos que partimos del líquido B puro y se añade líquido A gradualmente, manteniendo constante la temperatura en un valor T1. El sistema se encuentra inicialmente en el punto F (B puro) y se desplaza horizontalmente hacia la derecha. A lo largo de FC existe una sola fase, una disolución diluida del soluto A en el disolvente B. En el punto C se alcanza la solubilidad máxima del líquido A en el líquido B a T1 . Por lo tanto, al adicionar más A origina un sistema bifásico: la fase 1 es una disolución saturada de A en B, cuya composición es xA,1; la fase 2 es una disolución saturada de B en A, y su composición es xA,2 . La composición global del sistema bifásico en un punto cualquiera D de esta zona es xA,3 . En el punto D hay más cantidad de la fase 1 que de la fase 2. Si se sigue añadiendo más A, la composición global termina por alcanzar el punto E. En el punto E existe exactamente la cantidad de A necesaria para permitir que todo B se disuelva en A, formando una disolución saturada de B en A.
 y se desplaza horizontalmente hacia la derecha. A lo largo de FC existe una sola fase, una disolución diluida del soluto A en el disolvente B. En el punto C se alcanza la solubilidad máxima del líquido A en el líquido B a T1 . Por lo tanto, al adicionar más A origina un sistema bifásico: la fase 1 es una disolución saturada de A en B, cuya composición es xA,1; la fase 2 es una disolución saturada de B en A, y su composición es xA,2 . La composición global del sistema bifásico en un punto cualquiera D de esta zona es xA,3 . En el punto D hay más cantidad de la fase 1 que de la fase 2. Si se sigue añadiendo más A, la composición global termina por alcanzar el punto E. En el punto E existe exactamente la cantidad de A necesaria para permitir que todo B se disuelva en A, formando una disolución saturada de B en A..")
38
Por lo tanto, a partir de E el sistema vuelve a ser de una sola fase
Por lo tanto, a partir de E el sistema vuelve a ser de una sola fase. Desde E hasta H sólo se diluye la disolución de B en A. Para alcanzar el punto H es necesario una cantidad infinita de A. Con dos componentes y dos fases en equilibrio, el número de grados de libertad es 2. Sin embargo, como tanto P como T son constantes a lo largo de la línea CE, =0 en CE. Dos puntos que están sobre CE poseen valores idénticos para P, T , xA,1 , xA,2 , xB,1 y xB,2 . Al aumentar la temperatura, la zona de inmiscibilidad líquido-líquido disminuye hasta que se anula al alcanzar la temperatura crítica de la disolución (Tc). Por sobre Tc, los líquidos son totalmente miscibles. .
. Por sobre Tc, los líquidos son totalmente miscibles. .")
39
Temperatura superior de cosolubilidad
Temperatura inferior de cosolubilidad Para algunos pares de líquidos, como el agua con trietilamina una disminución de la temperatura conduce a una mayor miscibilidad. Algunas veces el sistema muestra una combinación de los comportamientos de los dos casos descritos anteriormente, como ocurre con nicotina-agua y m-toludina-glicerol

40
REGLA DE LA PALANCA Estas cantidades normalmente se expresan como porcentaje del peso (% peso) O EN MOLES, es una regla matemática valida para cualquier diagrama binario. Para calcular las cantidades de cada líquido, se construye una palanca sobre la isoterma con su punto de apoyo en la composición original de la mezcla (punto dado). El brazo de la palanca, opuesto a la composición de la fase cuya cantidad se calcula se divide por la longitud total de la palanca, para obtener la cantidad de dicha fase. Para un sistema bifásico con dos componentes, sean nA, n1, n2, el número de moles de A, el número de moles de la fase 1 y el número de moles de la fase 2 y vamos a deducir una relación entre la posición del punto D en la línea de conjunción CDE, con los valores diferentes de la fracción molar global xA,3 pero los mismos valores xA,1 y xA,2 .
 O EN MOLES, es una regla matemática valida para cualquier diagrama binario. Para calcular las cantidades de cada líquido, se construye una palanca sobre la isoterma con su punto de apoyo en la composición original de la mezcla (punto dado). El brazo de la palanca, opuesto a la composición de la fase cuya cantidad se calcula se divide por la longitud total de la palanca, para obtener la cantidad de dicha fase. Para un sistema bifásico con dos componentes, sean nA, n1, n2, el número de moles de A, el número de moles de la fase 1 y el número de moles de la fase 2 y vamos a deducir una relación entre la posición del punto D en la línea de conjunción CDE, con los valores diferentes de la fracción molar global xA,3 pero los mismos valores xA,1 y xA,2 .")
41
La fracción molar global de A
Además: Igualando estas dos expresiones:

42
EQUILIBRIO DE FASES EN SISTEMAS MULTICOMPONENTES
Equilibrio de fases sólido-líquido en sistemas de dos componentes Llamaremos disolvente al componente más abundante (B) y soluto al menos abundante (A) Cuando enfriamos una disolución a P=cte se produce la solidificación A + B (líquido) A (sólido) + disolución sobresaturada A + B (líquido) B (sólido) Punto de solidificación de la disolución
 y soluto al menos abundante (A) Cuando enfriamos una disolución a P=cte se produce la solidificación. A + B (líquido) A (sólido) + disolución sobresaturada. A + B (líquido) B (sólido) Punto de solidificación de la disolución.")
43
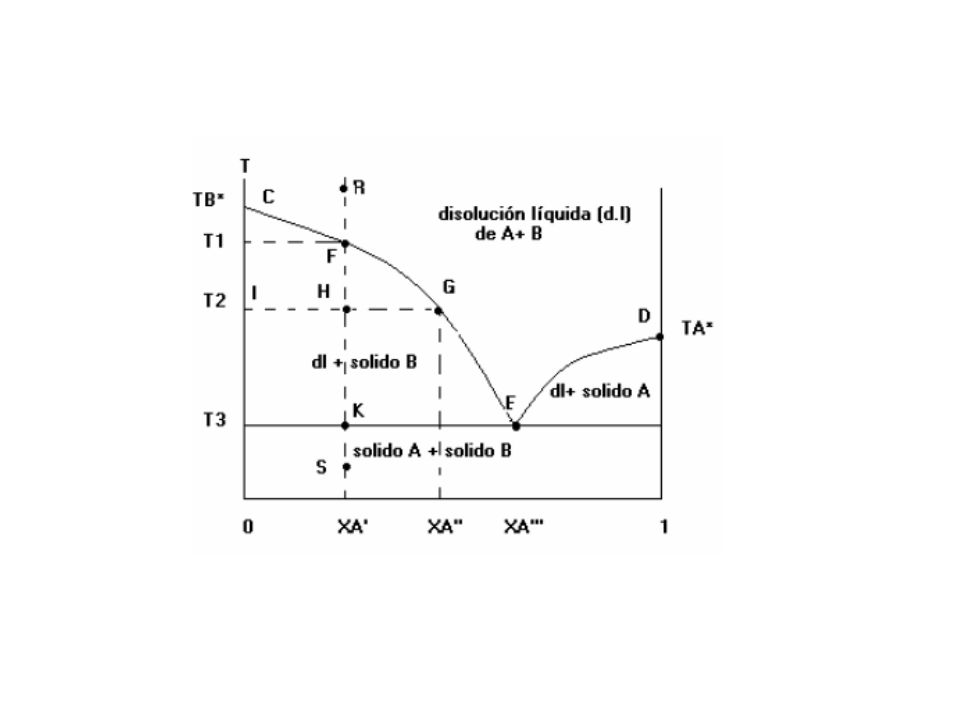
Bliquido = Bsolido D T1 C T2 I E eutéctico Ts única
xb´´´ xb´´ xb´ fig: diagrama de fases sólido-líquido cuando existe miscibilidad total en fase líquida e inmiscibilidad en fase sólida.

45
El efecto de la presión sobre los sólidos y los líquidos es pequeño, y a menos que se esté interesado en fenómenos que ocurran a presiones elevadas, se mantiene P constante a 1 atm. y se estudia el diagrama de fases sólido-líquido T-xA. Sean A y B dos sustancias miscibles en todas proporciones en la fase líquida y completamente inmiscibles en fase sólida. La mezcla de cantidades arbitrarias de los líquidos A y B origina un sistema monofásico que es una disolución de A y B. Como los sólidos A y B son completamente insolubles entre si, el enfriamiento de la disolución líquida de A y B ocasiona que A o B se congelen, abandonando la disolución. Una forma común de este tipo de diagramas es el de la figura anterior, donde TA* y TB* son los puntos de congelación de A puro y B puro. En el límite de baja temperatura, existe una mezcla bifásica del sólido puro A y el sólido puro B, ya que los sólidos son inmiscibles. En el límite de temperatura elevada existe una única fase formada por la disolución líquida de A y B, ya que los líquidos son miscibles

46
Viendo el enfriamiento de una disolución de A y B cuya concentración xA sea cercana a 1 (lado derecho del diagrama). Se alcanza una temperatura a la que el disolvente A comienza a congelarse, originando una región de dos fases con A sólido en equilibrio con una disolución líquida de A y B. La curva DE da el descenso del punto de congelación de A por la presencia del soluto B. Similarmente, si se enfría una disolución líquida con xB cercano a 1 (lado izquierdo del diagrama) provocará que la sustancia B pura se congele, por lo que CFGE es la curva de descenso del punto de congelación de B por el soluto A. Si se enfría una muestra bifásica formada por la disolución y cualquiera de los sólidos, la disolución se congelará completamente originando una mezcla de sólido A y sólido B. Las dos curvas del punto de congelación se interceptan en el punto E. Para una disolución con xA a la izquierda de E, se congelará el sólido B cuando T disminuye; si xA está a la derecha de E, será el sólido A el que se congele
 provocará que la sustancia B pura se congele, por lo que CFGE es la curva de descenso del punto de congelación de B por el soluto A. Si se enfría una muestra bifásica formada por la disolución y cualquiera de los sólidos, la disolución se congelará completamente originando una mezcla de sólido A y sólido B. Las dos curvas del punto de congelación se interceptan en el punto E. Para una disolución con xA a la izquierda de E, se congelará el sólido B cuando T disminuye; si xA está a la derecha de E, será el sólido A el que se congele.")
47
El punto E es el punto eutéctico (“que funde con facilidad”)
El punto E es el punto eutéctico (“que funde con facilidad”). Para los valores T y xA correspondientes al punto E, los potenciales químicos de A y B en la disolución son iguales a los de A y B puros, y tanto A como B se congelan cuando se enfría una disolución con la composición eutéctica xA´´´. Si se parte del punto R de la figura, y se enfría isobáricamente una disolución de A y B de composición xA´. El sistema es cerrado, por lo que la composición global permanece constante si se desciende verticalmente desde el punto R. Cuando T alcanza T1, empieza a aparecer el sólido B separado de la disolución Cuando B se congela, el valor de xA,l aumenta y (como A es ahora el soluto) el punto de congelación disminuye aún más. A una temperatura T2, existe un equilibrio entre una disolución cuya composición viene dada como xA´´ por el punto G y el sólido B, cuya composición viene dada por el punto I con xA=0. Los extremos de la línea de conjunción (GHI) dan las composiciones de las dos fases en equilibrio.
. Para los valores T y xA correspondientes al punto E, los potenciales químicos de A y B en la disolución son iguales a los de A y B puros, y tanto A como B se congelan cuando se enfría una disolución con la composición eutéctica xA´´´. Si se parte del punto R de la figura, y se enfría isobáricamente una disolución de A y B de composición xA´. El sistema es cerrado, por lo que la composición global permanece constante si se desciende verticalmente desde el punto R. Cuando T alcanza T1, empieza a. aparecer el sólido B separado de la disolución Cuando B se congela, el valor de xA,l aumenta y (como A es ahora el soluto) el punto de congelación disminuye aún más. A una temperatura T2, existe un equilibrio entre una disolución cuya composición viene dada como xA´´ por el punto G y el sólido B, cuya composición viene dada por el punto I con xA=0. Los extremos de la línea de conjunción (GHI) dan las composiciones de las dos fases en equilibrio.")
48
Si T continúa disminuyendo se alcanza finalmente la temperatura eutéctica T3 en el punto K. En ese punto la disolución tiene una composición xA’’’(punto E), y tanto A como B se congelan, por lo que ambos sólidos aparecen cuando se enfría una disolución de composición eutéctica. El resto de la disolución se congela también a la temperatura T3. En el punto K existen tres fases en equilibrio (disolución, sólido A y sólido B), por lo que no se puede aplicar la regla de la palanca. Con tres fases se tiene 1 grado de libertad (= 2- 3+2=1), este grado de libertad se elimina si se considera P constante e igual a 1atm. Por lo que no existen grados de libertad para el sistema trifásico, y la temperatura T3 permanece constante hasta que toda la disolución se haya congelado y el número de fases se reduzca a 2. Por debajo de T3 se enfría una mezcla de sólido A y sólido B (punto S). Si se invierte el proceso y se parte desde el punto S con A y B sólidos, el primer líquido que se forma tendrá la composición eutéctica xA’’’.
, este grado de libertad se elimina si se considera P constante e igual a 1atm. Por lo que no existen grados de libertad para el sistema trifásico, y la temperatura T3 permanece constante hasta que toda la disolución se haya congelado y el número de fases se reduzca a 2. Por debajo de T3 se enfría una mezcla de sólido A y sólido B (punto S). Si se invierte el proceso y se parte desde el punto S con A y B sólidos, el primer líquido que se forma tendrá la composición eutéctica xA’’’.")
49
El sistema se mantiene en el punto K hasta que se haya fundido todo A más la cantidad suficiente de B necesaria para formar una disolución de composición eutéctica. Luego, el sólido B restante se funde entre las temperaturas T3 y T1. Una mezcla sólida que posea la composición eutéctica fundirá completamente a una sola temperatura, la T3. Una disolución de A y B con la composición eutéctica congela a la temperatura T3. Pero, una mezcla eutéctica no es un compuesto , sino una mezcla íntima de cristales de A y cristales de B. Los sistemas que presentan un diagrama de fase como el antes descrito se llaman sistemas eutécticos simples. Algunos de ellos son los sistemas Pb-Sb, benceno-naftaleno, Si-Al, KCl-AgCl, Bi-Cd, C6H6-CH3Cl y cloroformo-anilina.

50
Para conseguir una idea aproximada de la forma de las curvas DE y CE, se desprecia la dependencia de la temperatura de ΔH fus, A y ΔH fus, B. Las ecuaciones que incluyen aproximaciones para las curvas DE y CE, son : R ln (xB)= ΔH fus, B ( 1/ TB* - 1/T) para CE R ln (xB)= ΔH fus, B ( 1/ TB* - 1/T) para DE
= ΔH fus, B ( 1/ TB* - 1/T) para CE. R ln (xB)= ΔH fus, B ( 1/ TB* - 1/T) para DE.")
51
Miscibilidad en fase líquida y en fase sólida
fig: diagrama de fases sólido-líquido del sistema Cu-Ni.

52
fig: diagrama de fase sólido-líquido T-xA para los sistemas a) Cu-Au, b) d-carvoxima-
l-carvoxima

53
Miscibilidad en fase líquida y miscibilidad parcial en fase sólida
fig: diagrama de fases sólido-líquido para el sistema Cu-Ag.

54
Formación de compuestos-miscibilidad en fase líquida e inmiscibilidad en fase sólida:
fig: diagrama de fases sólido líquido del sistema fenol-anilina. Los símbolos F(s), FA(s) y A(s) se refiere al fenol sólido, al compuesto de adición sólido y a la anilina sólida,respectivamente
, FA(s) y A(s) se refiere al fenol sólido, al compuesto de adición sólido y a la anilina sólida,respectivamente.")
55
Formación decompuestos con fusión incongruente-miscibilidad en fase líquida e inmiscibilidad en fase sólida fig: aparición de un punto peritéctico

56
EQUILIBRIO DE FASES EN SISTEMAS MULTICOMPONENTES
Equilibrio líquido-gas de mezclas binarias Relaciones presión-composición y temperatura-composición de una disolución ideal Sean dos líquidos A y B que forman una disolución ideal. Se mantiene la temperatura fija en un valor T por encima de los puntos de congelación de A y de B. Se representará la presión P del sistema frente a xA, la fracción molar de uno de los componentes. xA es la fracción molar global de A en el sistema, es decir: donde nA,l y nA,v son el número de moles de A en la fase líquida y vapor, respectivamente.Para un sistema cerrado, xA es constante, aunque nA,l y nA,v pueden variar.

57
Suponiendo que el sistema se encuentra en el interior de un cilindro cerrado por un pistón, que a su vez está inmerso en un baño termostático. En un instante inicial se supondrá que la presión externa es tan elevada como para que el sistema sea totalmente líquido (punto C). Al disminuir la presión por debajo de ese punto, se alcanza un valor tal que el líquido comienza a evaporarse (punto D). En ese punto el líquido posee una composición xA,l que es igual a xA, ya que sólo se ha evaporizado una cantidad infinitesimal de líquido. ¿Cual es la composición del primer vapor que aparece? La ley de Raoult, relaciona las fracciones molares de la fase vapor con la composición del líquido a través de:

58
Donde PA* y PB* son las presiones de vapor de los líquidos puros A y B a la temperatura T, la presión P del sistema es igual a PA +PB y se ha supuesto que el vapor es ideal. A partir de la relación anterior se llega a: Disolución ideal Sea A el componente más volátil, lo que significa que PA*>PB*. En este caso, la ecuación anterior queda como: xA,v/ xB,v > xA,l/ xB,l .

59
El vapor en equilibrio con una disolución ideal está más enriquecido en el componente más volátil que el líquido. Las relaciones anteriores son válidas para cualquier presión en la que se produzca el equilibrio líquido-vapor. Si se va disminuyendo la presión en forma isotérmica por debajo del punto D, haciendo que se evapore más líquido. Finalmente se alcanza el punto F, en el que se evaporiza la última gota de líquido. Para los puntos que están entre los puntos D y F, la fase líquida y gaseosa coexisten en equilibrio. Notar que como es un sistema cerrado, el valor de xA permanece constante a lo largo del proceso. Se puede repetir este experimento muchas veces pero a distintas composición. Luego se unen los puntos D y F que se consiguen en cada caso y se obtiene una representación . ¿Cuál es la ecuación de la curva D´s? Para los puntos D’s se obtiene una recta. Esto viene dado porque en esos puntos el líquido de composición xA,l está empezando a evaporarse, entonces la presión de vapor del líquido es:

60
C Disolución ideal Esta ec. es de una línea recta que se inicia en el valor de P*B cuando xA,l es cero y termina en P*A cuando xA,l vale 1. A lo largo de toda la línea DD´D´´ el líquido esta empezando a vaporizarse, por lo que la fracción molar global es xA es igual a la fracción molar de A en el líquido, xA,l. D´´ D´ D F´´ F´ F La línea DD´D´´ es la representación de la P total frente a la composición que es la fracción molar del líquido .

61
¿cuál es la ecuación de la curva FF´F´´?
La curva de las F’s es mas complicada. A lo largo de ella, la última gota del líquido se está evaporando, por lo que xA global será igual a xA,v, la fracción molar de A en el vapor. Por lo tanto la curva de F’s es una representación de la presión de vapor total P frente a xA,v. De la ley de Raoult y sustituyendo el valor de P de la relación para los puntos D, se obtiene: Y despejando xA,l Sustituyendo esta última expresión en P de la ley de Raoult, se llega a la ecuación de la presión: Disolución ideal Esta es la ecuación de P vs. xA,v y corresponde a la curva para F’s.

62
Entonces la curva superior corresponde a la curva de P frente a xA,l y la inferior corresponde a la curva de P frente a xA,v fig: diagrama de fases líquido-vapor de presión frente a composición para una disolución ideal a T constante. La línea inferior representa la curva de P frente a xA,v mientras que la línea superior es la curva de P frente a xA,l

63
Consideremos de nuevo el proceso que se inicia en el punto C, donde P es lo suficientemente elevado como para que sólo exista líquido. Al ser un sistema cerrado xA permanece constante. Por lo tanto, viene representado por una línea vertical en el diagrama de P vs. xA . En el punto D, con una presión igual a PD, el líquido comienza a evaporarse, lo que se quiere saber en ese punto es la concentración de xA,v para que exista equilibrio entre líquido-vapor cuando la presión es igual a PD, ya que se sabe que xA,l es igual a xA. La curva inferior es una representación de la ecuación para los puntos F’s y da P en función de xA,v o si lo veo al revés xA,v esta dado en función de P, por lo tanto para calcular xA,v para la presión PD debo ver el valor de la curva inferior para el cual P es igual a PD. Este es el punto G, y da la composición del vapor que aparece en primer lugar. Cuando la presión sigue disminuyendo, alcanza el punto PE. En el punto E el sistema está formado por dos fases, una fase líquida y otra fase vapor en equilibrio.

64
Para encontrar las composiciones de esa fase debo observar el valor que toma a esa presión la curva superior y la inferior para obtener los valores de xA,l (punto H) y xA,v (punto I), respectivamente. Por último, el punto F, con presión PF, el líquido restante se evapora, en ese punto xA = xA,v y xA,l = xA,4. Por debajo de F sólo se tiene vapor de composición xA. Por lo tanto, mientras la presión disminuye y el líquido se evapora en un sistema cerrado, xA,l se reduce desde D hasta J. Esto es debido a que A es más volátil que B. De la misma forma, mientras el líquido se evapora, xA,v disminuye desde G hasta F. Esto se debe a que el líquido que se evapora más tarde es más rico en el componente B. Para los estados en que las fases líquido y vapor están presentes simultáneamente, la presión P del sistema es igual a la presión de vapor del líquido. Una línea a lo largo de la cual permanece constante la composición, por ejemplo, la línea CDEF se denomina isopleta

65
En resumen, el diagrama de fase líquido-vapor de P frente a xA a temperatura constante para dos líquidos que forman una disolución ideal presentan tres regiones. En cualquier punto por encima de las dos curvas sólo existe líquido. En cualquier punto por debajo de ambas curvas sólo existe vapor. En cualquier punto intermedio E entre las dos curvas existen dos fases: líquida, cuya composición viene dada por el punto H (xA,l = xA,3), y otra de vapor, cuya composición viene dada por el punto I(xA,v = xA,2). La composición global del sistema bifásico viene dada por el valor de xA en el punto E. La línea horizontal HEI se denomina línea de conjunción. La región de dos fases comprendidas entre las curvas del líquido y del vapor es una zona del diagrama de fases en la que es imposible la existencia de una fase homogénea única. Un punto de esta zona de dos fases da la composición global, y las composiciones de las dos fases en equilibrio vienen dadas por los puntos situados en los extremos de la línea de conjunción.
, y otra de vapor, cuya composición viene dada por el punto I(xA,v = xA,2). La composición global del sistema bifásico viene dada por el valor de xA en el punto E. La línea horizontal HEI se denomina línea de conjunción. La región de dos fases comprendidas entre las curvas del líquido y del vapor es una zona del diagrama de fases en la que es imposible la existencia de una fase homogénea única. Un punto de esta zona de dos fases da la composición global, y las composiciones de las dos fases en equilibrio vienen dadas por los puntos situados en los extremos de la línea de conjunción..")
66
Relación temperatura-composición
disolución ideal a presión constante: Se describirá de una forma análoga al caso anterior, entonces se omitirán algunos detalles. Se representará temperatura frente a xA, la fracción molar global de uno de los componentes. Si TA* y TB* son los puntos de ebullición normal de los líquidos A y B puros, suponiendo que la presión es constante e igual a 1 atm. La curva inferior da T en función de xA,l para un sistema con una fase líquida y otra de vapor en equilibrio, y es la curva del punto de ebullición de la disolución ideal. La curva superior da T en función de xA,v para un sistema en el que existe un equilibrio líquido-vapor. La curva del vapor esta por encima de la curva del líquido en el diagrama de T frente a xA ,

67
pero está por debajo de la curva del líquido en el diagrama de P frente a xA. Esto es debido a que la fase vapor está favorecida por una T elevada y una P baja. Si se calienta isobáricamente un sistema cerrado de composición xA, el vapor aparece por primera vez en el punto L. Conforme aumenta la temperatura y se va evaporando más líquido, éste se va enriqueciendo en el componente B, menos volátil y con mayor punto de ebullición. Finalmente, se alcanza el punto N, donde se evapora la última gota de líquido

68
El vapor que aparece en primer lugar cuando se evapora una disolución de composición xA tiene una fracción molar xA,v dada por el punto Q. Si se saca este vapor del sistema y se condensa, se obtiene un líquido de composición xA,1. La evaporación de este líquido da lugar a vapor de composición inicial xA,2 (punto R). Por lo tanto, condensando y reevaporando la mezcla de forma sucesiva se puede separar A de B. Este proceso se llama destilación fraccionada. Para dibujar las curvas del diagrama se parte de PA*(T)y PB*(T), las presiones de vapor de los líquidos A y B puros, que se conocen en función de la temperatura (Diagramas de fase para 1 componente). Sea P* el valor constante de la presión , entonces P* = PA + PB, siendo PA y PB las presiones parciales de A y B en el vapor. La ley de Raoult dice O bien Disolución ideal
y PB*(T), las presiones de vapor de los líquidos A y B puros, que se conocen en función de la temperatura (Diagramas de fase para 1 componente). Sea P* el valor constante de la presión , entonces P* = PA + PB, siendo PA y PB las presiones parciales de A y B en el vapor. La ley de Raoult dice. O bien. Disolución ideal.")
69
Como PA*(T)y PB*(T) son funciones conocidas de la temperatura, se puede utilizar la relación anterior para calcular xA,l a cualquier T dada y de este modo dibujar la curva inferior (la del líquido). Para representar la curva del vapor, se utiliza xA,v=PA/P*=xA,lPA*/P* sustituyendo el valor de xA,l dado anteriormente se obtiene: Esta es la ecuación para xA,v en función de T.

70
Disoluciones no ideales:
Los diagramas de fases en los sistemas no ideales se obtienen midiendo la presión y la composición del vapor en equilibrio con un líquido de composición conocida. Si la disolución es ligeramente no ideal, las curvas se parecen a las de las disoluciones ideales, y no existen cambios significativos. En cambio, si la disolución presenta una desviación considerable del comportamiento ideal como para que exista un máximo o un mínimo en la curva de P frente a xA,l se produce la aparición de un fenómeno nuevo. En el caso de que ocurra un máximo en la curva superior del diagrama de fases de P frente a xA ,(desviación positiva de la ley de Raoult) es decir, un máximo de la curva de P frente a xA,l. ¿Qué forma tendrá la curva inferior?. La curva inferior necesariamente tendrá un máximo también en ese punto.
 es decir, un máximo de la curva de P frente a xA,l. ¿Qué forma tendrá la curva inferior . La curva inferior necesariamente tendrá un máximo también en ese punto.")
71
Esto se debe a que si se reduce isotérmamente la presión, alcanzamos el punto donde el líquido comienza a evaporarse, para conocer la composición del primer vapor que aparece se necesita el valor de xA,v que corresponde a la presión en el punto donde el líquido comienza a evaporarse, si la curva inferior no presentase también un máximo en ese punto no se podría conocer debido a que no existiría ningún punto donde la curva inferior presentase esa presión, por lo tanto no tendría sentido. Entonces, en el punto donde la curva superior presenta un máximo, la curva inferior también debe tener un máximo. Entonces el diagrama puede ser como:

72
Desviaciones de la ley de Raoult
Desviaciones positivas Desviaciones negativas Interacciones intermoleculares < que interacciones en el líquido puro Interacciones intermoleculares > que Interacciones en líquido puro

73
Los diagramas líquido-gas de las disoluciones no ideales son mucho
más complejos. Encontrándose mínimos o máximos. Azeótropos Mezcla cloroformo-acetona No se pueden separar

74
Destilación

75
DIAGRAMAS DE FASE DE 3 COMPONENTES O TERNARIO
En este tipo de sistemas se tienen 4 variables independientes: presión, temperatura y dos concentraciones. En materiales cerámicos, dada la naturaleza y estabilidad de los compuestos con que habitualmente se trabaja, es posible debido a sus bajas presiones de vapor, despreciar el efecto de la presión en el estudio de diagramas de equilibrio de fases, de tal forma que la relación que da cuenta del fenómeno queda: =3-F+2=5-F Donde: 4 fases: univariante ( =1) 3 fases: bivariante ( =2) 2 fases: trivariante ( =3) 1 fase = cuatrivariante ( =4)
 3 fases: bivariante ( =2) 2 fases: trivariante ( =3) 1 fase = cuatrivariante ( =4)")
76
Para realizar un diagrama bidimensional es necesario mantener constantes 2 variables, P y T Se eligen por ej como variables las fracciones molares de A y B y queda fijada la de C. Al requerirse 3 variables para expresar el equilibrio heterogéneo de un sistema ternario, la representación gráfica necesaria debe ser tridimensional. Si se toma el plano x-y para graficar las concentraciones, y la coordenada z para expresar las temperaturas, se tendrá una representación como:

77
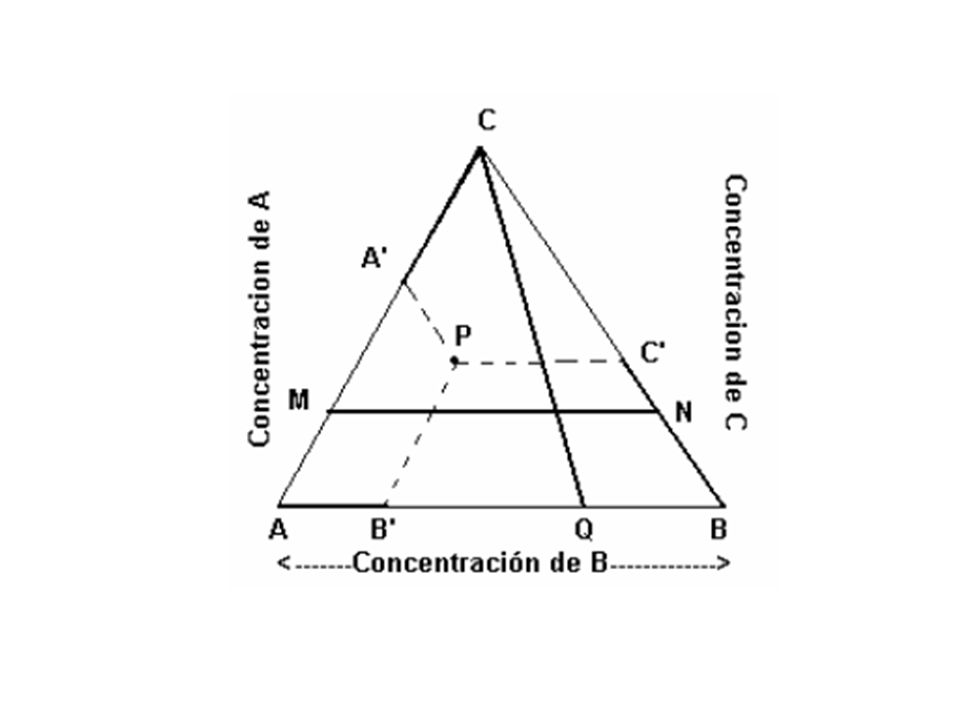
Pero por sus propiedades geométricas, un triángulo equilátero es mucho más cómodo para representar concentraciones. El triángulo equilátero elegido para representar las concentraciones de un sistema ternario tiene la enorme ventaja de ser una figura muy regular con bastante simetría, y con una geometría muy simple. A continuación se verán las propiedades geométricas que se usan al estudiar el equilibrio heterogéneo en sistemas ternarios. 1) El triángulo equilátero tiene iguales sus lados, sus ángulos internos y externos, sus alturas, sus transversales de gravedad y sus bisectrices. 2) Si los lados del triángulo expresan las concentraciones de A, B y C (en fracciones molares o en porcentaje en peso), entonces la concentración de A, B y C de un punto P cualquiera en el interior del triángulo viene dada por:
 El triángulo equilátero tiene iguales sus lados, sus ángulos internos y externos, sus alturas, sus transversales de gravedad y sus bisectrices. 2) Si los lados del triángulo expresan las concentraciones de A, B y C (en fracciones molares o en porcentaje en peso), entonces la concentración de A, B y C de un punto P cualquiera en el interior del triángulo viene dada por:")
78
AB’= xB ( o porcentaje de B);
BC’ = xC ( o porcentaje de C); CA’ = xA ( o porcentaje de A); Si el punto P está expresado en coordenadas dadas en porcentaje en peso, no tiene porqué coincidir con el punto P equivalente, expresado en coordenadas dadas en fracciones molares. 3)Una transversal cualquiera, por ejemplo CQ en la figura, es el lugar geométrico de los puntos que cumplen la condición xA/ xB= constante, o bien %A/ %B= constante, en el caso que el triángulo esté expresado en porcentaje en peso. 4)Una paralela a cualquier lado del triángulo, por ejemplo MN / AB en la figura, debe satisfacer la relación que la suma de las concentraciones de los componentes ubicados en el lado paralelo es constante. Así, para MN se tiene xA +xB = 1 – xC =constante, o bien (%A +%B)=100-%C = constante
; CA’ = xA ( o porcentaje de A); Si el punto P está expresado en coordenadas dadas en porcentaje en peso, no tiene porqué coincidir con el punto P equivalente, expresado en coordenadas dadas en fracciones molares. 3)Una transversal cualquiera, por ejemplo CQ en la figura, es el lugar geométrico de los puntos que cumplen la condición xA/ xB= constante, o bien %A/ %B= constante, en el caso que el triángulo esté expresado en porcentaje en peso. 4)Una paralela a cualquier lado del triángulo, por ejemplo MN / AB en la figura, debe satisfacer la relación que la suma de las concentraciones de los componentes ubicados en el lado paralelo es constante. Así, para MN se tiene xA +xB = 1 – xC =constante, o bien (%A +%B)=100-%C = constante.")
80
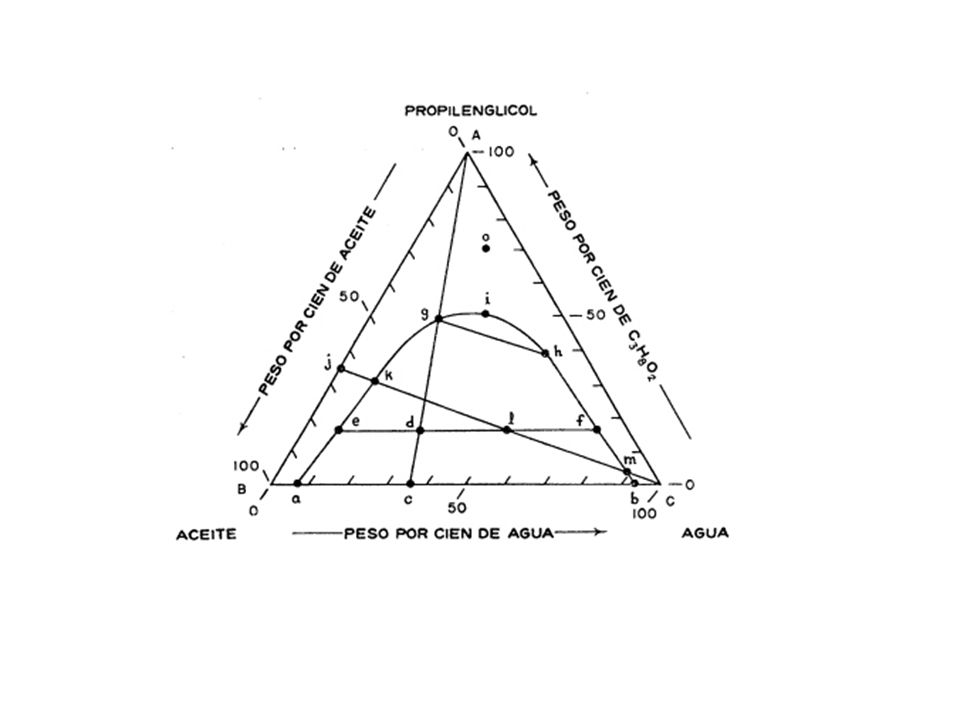
5) Si se elige, por ejemplo el punto P ubicado en el interior del triángulo, las concentraciones de él pueden quedar expresadas en términos de A´B´C. 6) El vértice opuesto a cada lado corresponde al cien por cien de dicho componente. Vamos a considerar un sistema de tres componentes: En primer lugar, una mezcla de aceite y agua (línea BC de la figura ), en la que el aceite y el agua son prácticamente inmiscibles, pero ambos se hacen miscibles con el agente solubilizante propilenglicol: Sólo puede añadirse una pequeña cantidad de agua al aceite antes de que la mezcla, parcialmente miscible, se separe formando dos capas. Cuando la composición total de la mezcla es c, o cualquier otro valor entre a y b, una de las disoluciones binarias conjugadas, o capas, contiene agua en aceite, con una composición a, y la otra contiene aceite en agua, con la composición b.
 El vértice opuesto a cada lado corresponde al cien por cien de dicho componente. Vamos a considerar un sistema de tres componentes: En primer lugar, una mezcla de aceite y agua (línea BC de la figura ), en la que el aceite y el agua son prácticamente inmiscibles, pero ambos se hacen miscibles con el agente solubilizante propilenglicol: Sólo puede añadirse una pequeña cantidad de agua al aceite antes de que la mezcla, parcialmente miscible, se separe formando dos capas. Cuando la composición total de la mezcla es c, o cualquier otro valor entre a y b, una de las disoluciones binarias conjugadas, o capas, contiene agua en aceite, con una composición a, y la otra contiene aceite en agua, con la composición b.")
82
Las dos son disoluciones binarias, puesto que contienen dos componentes: agua y aceite. La adición de propilenglicol, agente solubilizante, o de unión, pues es miscible con ambos componentes, produce un aumento de la solubilidad mutua del agua y el aceite. La línea cA señala las variaciones totales de propilenglicol en la composición. Cuando se ha añadido la cantidad suficiente de este compuesto para que la composición total sea d, las dos disoluciones ternarias conjugadas inmiscibles son e y f y cada una de las dos contiene agua, aceite y propilenglicol. Estas dos capas son disoluciones ternarias, porque cada una de ellas consta de tres componentes. La línea ef, denominada línea de unión o de enlace, es paralela a la base del triángulo solamente si el propilenglicol se distribuye de una manera uniforme entre el aceite y el agua. Sin embargo, éste no es el caso general de los agentes solubilizantes, ya que éstos no se reparten en cantidades iguales en las dos capas líquidas, sino que lo hacen de acuerdo con el coeficiente de reparto del agente en los otros dos líquidos.

83
Las composiciones de las disoluciones inmiscibles y, por tanto, las posiciones de las líneas de unión, han de ser determinadas experimentalmente. En el punto g las dos capas ternarias tienen las composiciones g y h, y cuando se añade más propilenglicol la capa h desaparece, y se alcanza una disolución homogénea. En todos los puntos que están fuera de la curva existe una sola fase, mientras que en los que están debajo de la misma, se forman dos fases separadas. Las composiciones de las dos fases sólo son idénticas en el punto i en el cual desaparecen las líneas de unión y las dos capas se funden en una sola.

Presentaciones similares




>")




>")









