Descargar la presentación
La descarga está en progreso. Por favor, espere

1
Dr. Héctor Cuevas y Dra. Edna Aizpuru
SINDROMES GENETICOS PEDIATRIA Samuel Almeida Navarro MIP Coordinadores: Dr. Héctor Cuevas y Dra. Edna Aizpuru

2
SINDROMES GENETICOS DEFINICION
Las enfermedades genéticas corresponden a un grupo heterogéneo de afecciones que en su etiología presentan un significativo componente genético. Ello puede ser alguna alteración monogénica, multifactoriales o en cromosomas.

3
SINDROMES GENETICOS El nacimiento de un niño con una enfermedad genética, es habitualmente un evento inesperado, muy angustioso para los padres y la familia. Por esta razón el equipo médico debe estar preparado para hacerse cargo en forma rápida y eficiente del niño y de sus familiares.

4
SINDROMES GENETICOS Un diagnóstico oportuno permitirá por una parte, evaluar la situación, intentar aproximarse a un diagnóstico específico y en lo posible, a una terapia adecuada, y, por otra parte, orientar y dar apoyo a los padres y en caso necesario, entregar un consejo genético apropiado.

5
¿En caso de tener otro hijo es probable que nazca igual?
SINDROMES GENETICOS Es por esto que un pediatra al igual que un medico general siempre se enfrenta a algunas preguntas clásicas de los padres: ¿Porqué ocurrió? ¿En caso de tener otro hijo es probable que nazca igual?

6
SINDROMES GENETICOS Una historia clinica adecuada es necesaria para un acertado diagnostico en los Sx. Geneticos, en ella siempre debe de incluir: Edad de los padres al momento de la gestacion Abortos u obitos en la familia Familiares con retraso mental, malformaciones y/o sindromes geneticos Consanguineidad, endogamia Religión de los padres

7
SINDROMES GENETICOS CONCEPTOS BASICOS

8
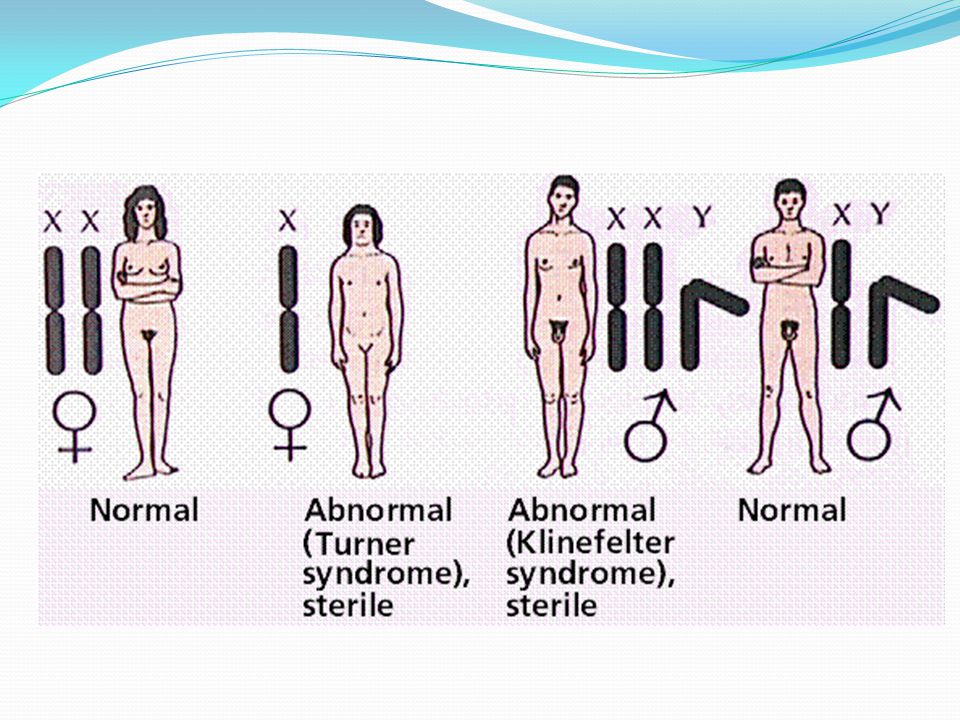
SINDROMES GENETICOS AUTOSOMICO DOMINANTE
Si simplemente uno de los padres tiene un gen defectuoso dominante, CADA hijo tiene un 50% de probabilidades de heredar el trastorno.

9
SINDROMES GENETICOS AUTOSOMICO RECESIVO
La herencia recesiva significa que AMBOS genes de un par deben estar defectuosos para causar la enfermedad

10
LIGADO A CROMOSOMAS SEXUALES
SINDROMES GENETICOS LIGADO A CROMOSOMAS SEXUALES Las enfermedades ligadas al sexo se heredan a través de uno de los "cromosomas sexuales" X o Y.

12
SINDROMES GENETICOS

13
PREVALENCIA E INCIDENCIA
SINDROMES GENETICOS PREVALENCIA E INCIDENCIA La prevalencia en LA en hospitales pediátricos es de 62.5 % y su incidencia de 17 %. Se estima que aprox. el 3-7% de la población presenta un problema genético En los países desarrollados, se ha estimado que 52.8 % de ingresos a hospitales pediátricos presentan alguna patología genética Santos, M. "Apuntes de Genética General, Humana y Médica". Editado por la Fac. Ciencias Biológicas, P. Universidad Católica de Chile, 2000.

14
TIPO DE ENFERMEDAD GENETICA
SINDROMES GENETICOS PREVALENCIA E INCIDENCIA Los SG corresponden a un aproximado 2/3 de las defunciones hospitalarias en Hospitales pediatricos en EU. TIPO DE ENFERMEDAD GENETICA PREVALENCIA POR 1000 RN AUTOSOMICA DOMINANTE AUTOSOMICA RECESIVA LIGADAS AL X AFECCIONES CROMOSOMICA MALFORMACIONES CONGENITAS Santos, M. "Apuntes de Genética General, Humana y Médica". Editado por la Fac. Ciencias Biológicas, P. Universidad Católica de Chile, 2000.

15
CASOS CLINICOS

16
Caso 1:

17
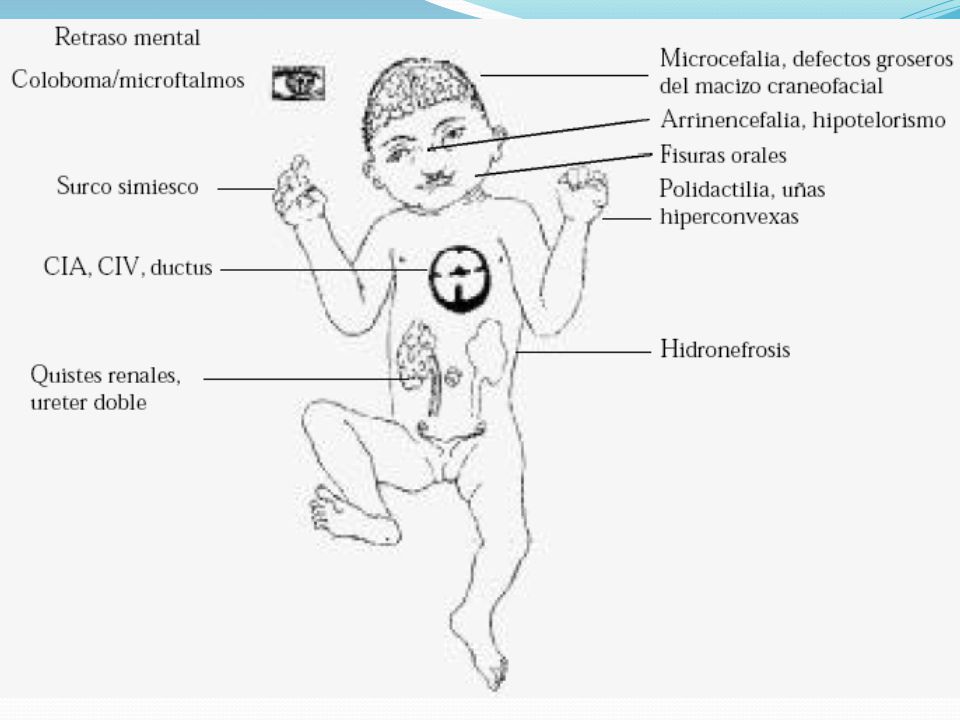
SINDROME DE PATAU Se trata de la trisomía menos frecuente, descubierta en 1960 por Patau. Resulta de la presencia de un cromosoma 13 suplementario El cariotipo da 47 cromosomas y sirve de diagnóstico prenatal por amniocentesis o cordiocentesis.

18
SINDROME DE PATAU Los afectados mueren poco tiempo después de nacer, la mayoría a los 3 meses, como mucho llegan al año. Se suele asociar con un problema meiótico materno, más que paterno. El riesgo aumenta con la edad de la mujer.

19
SINDROME DE PATAU Anomalías en el sistema nervioso: Retraso mental, Holoprosencefalia, Dilatación de la bifurcación ventricular y alargamiento del surco posterior. Anomalías faciales: hipotelorismo que puede llegar a la presencia de un solo ojo y coloboma, Labio leporino y Trastornos en la lengua, aparición de más de dos Narices.

20
SINDROME DE PATAU Anomalías renales: Hidronefrosis y Aumento de tamaño del riñón Anomalías cardíacas: Comunicación interventricular, Displasia valvular y Tetralogía de Fallot Anomalías de miembros: Polidactilia y Pie vago. Anomalías en abdomen: Onfalocele y Extrofía vesicular Hipotonía muscular

22
Caso 2:

23
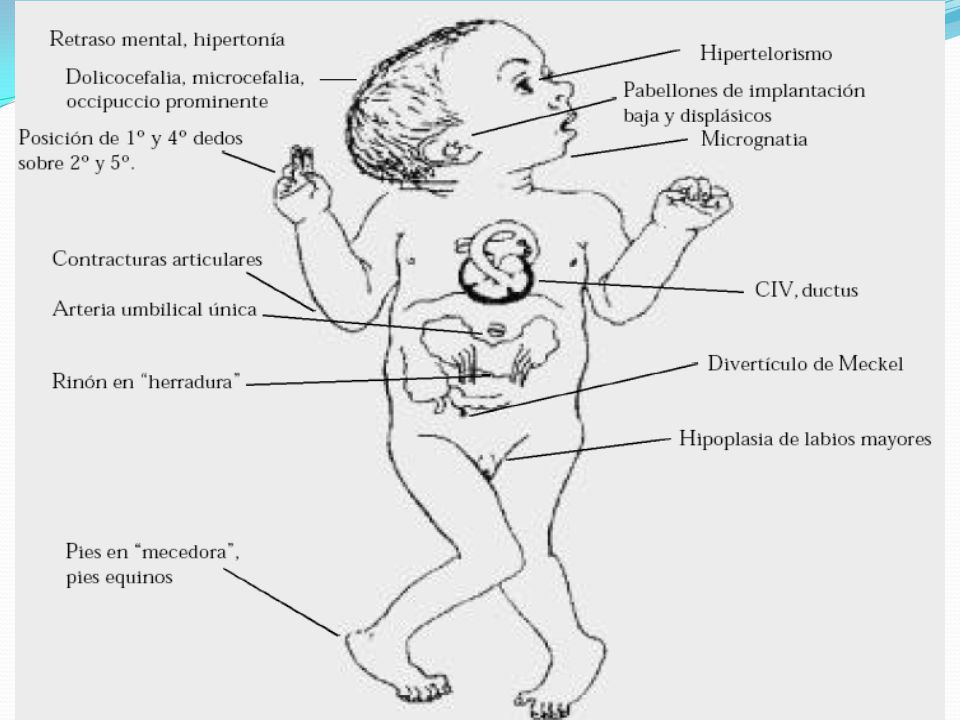
SINDROME DE EDWARDS Descrita por John H Edwards en la Universidad de Wisconsin Trisomia, translocacion o mosaicismo. Tasa de mortalidad en los recién nacidos, por sobre el 90% de los casos, no llegan a un año de vida. Escasos supervivientes. El 50% de los errores en la separación de los cromosomas en la ovogénesis, en meiosis II

24
SINDROME DE EDWARDS I. Dismorfismos II. Malformaciones Faciales
Músculo-Esqueléticos II. Malformaciones Anomalías del SNC y cráneo, faciales, cuello, cardiovasculares, gastrointestinales, genitourinarias, de extremidades, entre otras

25
SINDROME DE EDWARDS Su frecuencia se calcula de 1 en 4000 nacidos vivos. Con un mayor número de mujeres afectadas 2:1. Signos radiológicos: Esternón corto. Núcleos de osificación reducidos. Pelvis pequeñas. Luxación de Cadera.

26
SINDROME DE EDWARDS EVOLUCION
Mortalidad del 95% en el primer año de vida. La tasa de mortalidad en los supervivientes es del 2% a los 5 años de vida. Las niñas presentan mayor tasa de supervivencia. Causa principal de fallecimiento: Cardiopatía congénita, apneas, y neumonía.

28
Caso 3:

29
SINDROME DE DOWN Cromosomopatía más común.
65 a 80% de las gestaciones con trisomía 21 son abortos espontáneos. Frecuencia en R.N.: 1 / 700. Aneuploidia más común compatible con la vida.

30
SINDROME DE DOWN ETIOLOGIA Trisomía 21 Regular.
El 94% de los casos. Por no disyunción meiótica del óvulo. Trisomía por traslocación Robertsoniana. El 45 de los casos. Traslocación entre el cromosoma 21 y otro cromosoma acrocéntrico que normalmente es el 14 o el 22, ocasionalmente, 21. Mosaico, con cariotipo normal y trisomía 21. Un 1% de los pacientes presentan. La realización del cariotipo es obligada.

31
CARACTERISTICAS CLINICAS
SINDROME DE DOWN CARACTERISTICAS CLINICAS Hipotonía e hiperlaxitud ligamentosa. Desarrollo Psicomotor muy lento. Retardo Mental acentuado

32
CARACTERÍSTICAS CRANEOFACIALES:
SINDROME DE DOWN CARACTERISTICAS CLINICAS CARACTERÍSTICAS CRANEOFACIALES: Braquicefalia. Puente nasal deprimido. Occipital aplanado. Implantación baja de cabello. Cuello corto. Hipoplasia medio facial.

33
CARACTERISTICAS CLINICAS
SINDROME DE DOWN CARACTERISTICAS CLINICAS CARA: OJOS “Almendrados”. Fisuras palpebrales hacia arriba. Epicanto interno. Manchas de Brushfield. Estrabismo Nistagmos PABELLONES AURICULARES Helix muy plegado. Malformados y pequeños. Ausencia de lóbulos

34
CARACTERISTICAS CLINICAS
SINDROME DE DOWN CARACTERISTICAS CLINICAS CARA: Micrognatia. Paladar ojival. Macroglosia. MANOS: Pequeñas y cuadradas. Braquidactilia. Clinodactilia. hipoplasia de la falange media del 5º dedo. Surco palmar único.

35
CARACTERISTICAS CLINICAS
SINDROME DE DOWN CARACTERISTICAS CLINICAS PIES: 1º Espacio interdigital aumentado. Arco tibial en área halucal. Pliegue plantar entre el 1º y 2º ortejos.

36
CARACTERISTICAS CLINICAS
SINDROME DE DOWN CARACTERISTICAS CLINICAS GENITALES: Pene pequeño. Volumen testicular menor que el de los niños de su edad. Criptorquidia relativamente frecuente. PIEL: Es redundante en la región cervical (fetal y neonatal). Livedo reticularis de predominio en extremidades inferiores. Hiperqueratósica con el tiempo.
. Livedo reticularis de predominio en extremidades inferiores. Hiperqueratósica con el tiempo.")
37
CARACTERISTICAS CLINICAS
SINDROME DE DOWN CARACTERISTICAS CLINICAS Cardiopatía congénita: 45% Canal atrioventricular % CIV % CIA PCA Tetralogía de Fallot Gastrointestinales: 10 – 18%. Hernia Umbilical. Atresia duodenal. La enfermedad de Hirschsprung.

38
ENTIDADES CLINICAS ASOCIADAS
SINDROME DE DOWN ENTIDADES CLINICAS ASOCIADAS Infecciones recurrentes. Respuesta Inmunológica comprometida. Leucemia aguda linfoblástica (15 a 20 veces más frecuente). Hipotiroidismo (1 : 10). Hipoacusia conductiva o neurosensorial. Problemas visuales 50%. Inestabilidad atlanto-axoidea. La mitad de los niños fallecen entre los 3 y 4 años Alzheimer.
. Hipotiroidismo (1 : 10). Hipoacusia conductiva o neurosensorial. Problemas visuales 50%. Inestabilidad atlanto-axoidea. La mitad de los niños fallecen entre los 3 y 4 años. Alzheimer.")
39
SINDROME DE DOWN Las apneas obstructivas del sueño son frecuentes.
A pesar de que la mayoría de ciclos son anovulatorios pueden llegar a concebir. La erección y eyaculación completas son difíciles.

40
SINDROME DE DOWN DIAGNOSTICO
Las características fenotípicas. Hipotonía y llanto característico, agudo y entrecortado. Fenotipo característico. CARIOTIPO.

41
SINDROME DE DOWN Diagnóstico Prenatal:
Estudio citogenético de vellosidades coriónicas o de líquido amniótico. Amniocentesis temprana. 12-14 Semanas de Gestación. .

42
SINDROME DE DOWN RIESGO DE RECURRENCIA
Menor del 1% cuando es ocasionado por Trisomía regular. Del 10 a 15% cuando la madre es portadora de una traslocación. Del 2 al 5 % cuando el padre es portador de una traslocación. Traslocación Robertsoniana entre dos cromosomas 21 el riesgo de recurrencia es del 100% independientemente del sexo que lo transmita.

43
SINDROME DE DOWN RIESGO DE RECURRENCIA EDAD 20 20-24 25-29 30-34 35-39
40-44 >45 FRECUENCIA 1:1,150 1:1,400 1:1,100 1: 700 1: 275 1: 100 1:

44
Caso 4:

45
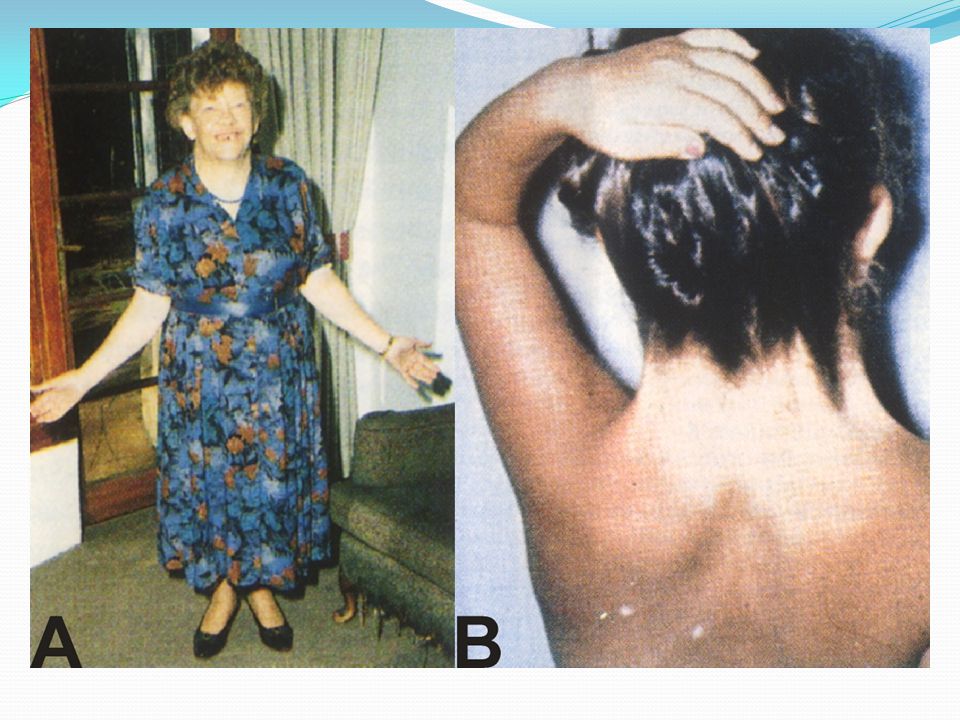
Resulta de la ausencia total o parcial de un segundo cromosoma sexual
SINDROME DE TURNER Resulta de la ausencia total o parcial de un segundo cromosoma sexual Características: talla baja, déficit cognitivo, disgenesia gonadal, infertilidad, displasia pabellones auriculares, micrognatia, cuello alado (higroma coli), implantación baja cabello, problemas cardiovasculares (aorta), cubitus valgus, alteraciones renales.
, implantación baja cabello, problemas cardiovasculares (aorta), cubitus valgus, alteraciones renales.")
46
SINDROME DE TURNER Incidencia: 1/2000-3000 recién nacidas.
Frecuencia de concepciones 45,X es mucho mayor y es muy común en abortos espontáneos del primer trimestre (99% de 45,X son abortados espontáneamente (??) Cariotipo: 45,X/46,XY /10, cuadro muy variable. Fenotipo femenino con signos de virilización.
 Cariotipo: 45,X/46,XY 1.5/10,000 cuadro muy variable. Fenotipo femenino con signos de virilización.")
48
CARIOTIPOS RELACIONADOS
-Monosomía del X (60% casos) ,X -Mosaicos (24% de los casos) ,X/46,XX 45,X/47,XXX 45,X/46,XY (4% casos) 45,X/46,XX/47,XXX Alteraciones estructurales isocromosoma Xq: 46,X,i(Xq) -pérdida Xp deleción: 46,X,del(Xp) -pérdida Xq deleción: 46,X,del(Xq) trans. X/A trans. X/X anillo X: 46,X,r(X) -deleción Yp ,X del (Yp) -isocromosoma Y ,X,i(Yq) Numéricas Estructurales
 45,X. -Mosaicos (24% de los casos) 45,X/46,XX. 45,X/47,XXX. 45,X/46,XY (4% casos) 45,X/46,XX/47,XXX. Alteraciones estructurales isocromosoma Xq: 46,X,i(Xq) -pérdida Xp deleción: 46,X,del(Xp) -pérdida Xq deleción: 46,X,del(Xq) trans. X/A. trans. X/X. anillo X: 46,X,r(X) -deleción Yp 46,X del (Yp) -isocromosoma Y 46,X,i(Yq) Numéricas. Estructurales.")
49
Caso 5:

50
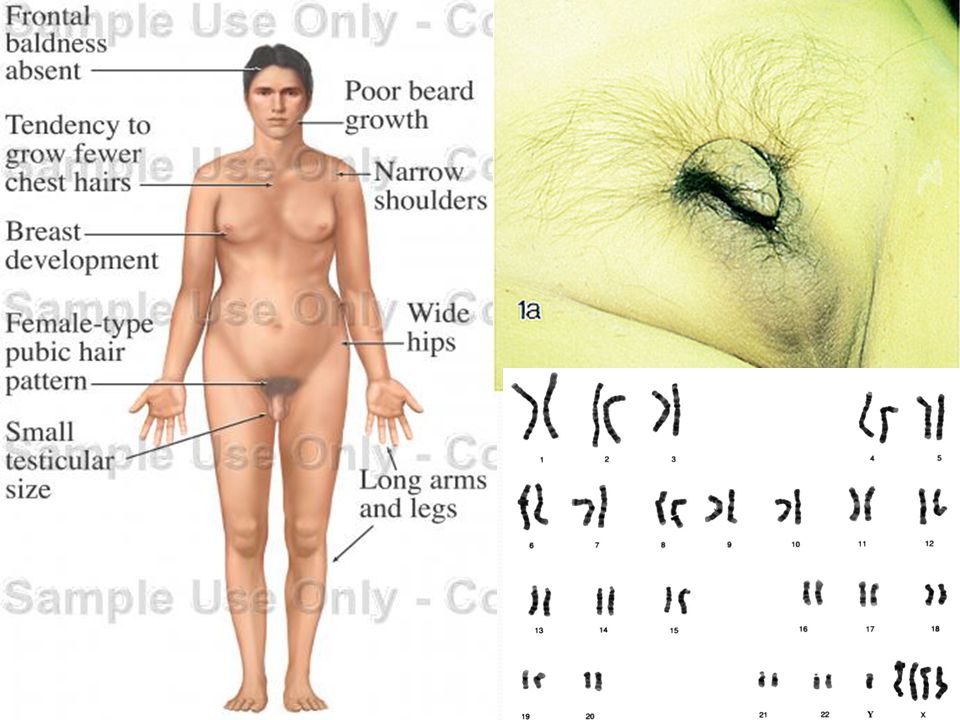
SINDROME DE KLINEFELTER
Descrito en 1942 por Klinefelter y col. Anomalía cromosomas sexuales más común en humanos. Varones con ginecomastia, testículos pequeños, azoospermia, vello púbico y facial escaso. Testosterona disminuída, longitud pene disminuído, distribución ginecoide de la grasa corporal.

52
Caso 6:

53
SINDROME DE NOONAN El síndrome de Noonan es causado por anomalías en los genes KRAS y PTPN11. Aproximadamente la mitad de las personas afectadas por este síndrome tienen una mutación en el gen PTPN11. Las personas con una anomalía en el gen KRAS presentan una forma atípica o severa del síndrome de Noonan

54
SINDROME DE NOONAN Similar a Turner, pero en mujeres.
Esta enfermedad ocurre en aproximadamente 1 de cada 1000 a 2500 niños. Autonómica dominante.

55
CARACTERISTICAS CLINICAS
SINDROME DE NOONAN CARACTERISTICAS CLINICAS Retardo en la pubertad Ojos de base amplia o inclinados hacia abajo Pérdida de la audición (varía) Orejas de implantación baja o de forma anormal Retardo mental leve (sólo en aproximadamente el 25% de los casos) Párpados caídos (ptosis) Estatura baja Pene pequeño Testículos no descendidos Forma inusual del tórax (generalmente un tórax hundido llamado tórax excavado) Cuello con pliegues y de apariencia corta
 Orejas de implantación baja o de forma anormal. Retardo mental leve (sólo en aproximadamente el 25% de los casos) Párpados caídos (ptosis) Estatura baja. Pene pequeño. Testículos no descendidos. Forma inusual del tórax (generalmente un tórax hundido llamado tórax excavado) Cuello con pliegues y de apariencia corta.")
56
Caso 7:

57
CRI-DU-CHAT Maullido de gato
El síndrome de Lejeune, es una alteración cromosómica provocada por deleción estructural de parte o de todo el brazo corto del cromosoma 5. Caracterizada por un llanto que se asemeja al maullido de un gato y que se va modificando con el tiempo. Descrito inicialmente por Lejeune en 1963. Tiene una prevalencia estimada de aproximadamente de 1/ nacimientos y predomina en las niñas

58
CRI-DU-CHAT Maullido de gato
El afectado normalmente presenta retraso de crecimiento intrauterino con peso bajo al nacimiento y llanto característico que recuerda al maullido de gato, por laringomalacia con hipoplasia de la epiglotis y relajación de los pliegues ariepiglóticos

59
CARACTERISTICAS CLINICAS
Cri-Du-Chat CARACTERISTICAS CLINICAS Bajo peso al nacimiento: aproximadamente 2 kg. Crecimiento lento Llanto característico: Hz, Normal: Hz Perímetro craneal reducido Deficiencia mental Hipotonía Marcado sentido del humor. Cariñosos y muy afectivos. Miedo a determinados objetos. Alteraciones a nivel psicofísico.

60
CRI-DU-CHAT Maullido de gato
La esperanza de vida no se puede evaluar con certeza, pero se describen casos de adultos con pocos casos de fallecimiento. Los problemas más graves se deben a defectos cardíacos y a las complicaciones respiratorias.

61
Caso 8:

62
Sx. WOLF-HIRSCHHORN Causada por una micro-deleción distal del brazo corto del cromosoma 4. Las principales características son los rasgosfaciales peculiares, crisis convulsivas y retardo psicomotor y del desarrollo. Su frecuencia depresentación es de 1 por cada 50,000 nacidos vivos, presentando el doble de frecuencia en mujeres Síndrome de Wolf-Hirschhorn: Microdelecióndistal del brazo corto del cromosoma 4: JORGE A. AVIÑA F.1, DANIEL A. HERNÁNDEZ A. Rev Chil Pediatr 2008; 79 (1): 50-53
:")
63
Sx. WOLF-HIRSCHHORN Los criterios diagnósticos son:
dismorfia facial característica en "yelmo grie-go", retardo del crecimiento y manifestacionesde déficit neurológico: retraso mental e hipotonía Convulsiones5. Síndrome de Wolf-Hirschhorn: Microdelecióndistal del brazo corto del cromosoma 4: JORGE A. AVIÑA F.1, DANIEL A. HERNÁNDEZ A. Rev Chil Pediatr 2008; 79 (1): 50-53
:")
64
Sx. WOLF-HIRSCHHORN Mutación de novo
Causa más frecuente el 85% de éstos son deleción proveniente del padre. 12% pueden tener alteraciones como un cromosoma 4 en anillo, mosaicismo, o traslocación esporádica no balanceada. El pronóstico es desfavorable a corto plazo, supervivencia es aproximadamente de 2 años de vida, muriendo por complicaciones respiratorias y cardíacas Sólo un tercio sobrevive un tiempo mayor Síndrome de Wolf-Hirschhorn: Microdelecióndistal del brazo corto del cromosoma 4: JORGE A. AVIÑA F.1, DANIEL A. HERNÁNDEZ A. Rev Chil Pediatr 2008; 79 (1): 50-53
:")
65
Caso 9:

66
SINDROME DE Di GEORGE Variabilidad clínica.
Se conoce como CATCH (defectos cardíacos, facies anormal, defectos sistema inmune (hipoplasia timo), paladar hendido, hipocalcemia) retardo mental moderado. Detectable citogenéticamente, comparte región con síndrome velocardio-facial.
, paladar hendido, hipocalcemia) retardo mental moderado. Detectable citogenéticamente, comparte región con síndrome velocardio-facial.")
67
SINDROME DE Di GEORGE Frecuencia: 1/3,000-4,000 RN
En 15% parejas con hijo afectado, uno de los padres tb tiene la deleción (50% riesgo recurrencia). En ausencia de deleción parental recurrencia es rara. Ecografía anormal (anomalía cardiaca) muestra 3/26 casos sin historia familiar positiva.
. En ausencia de deleción parental recurrencia es rara. Ecografía anormal (anomalía cardiaca) muestra 3/26 casos sin historia familiar positiva.")
68
Caso 10:

69
SX. TREACHER-COLLINS Ocurre en 1:10,000 Nacidos vivos
Autosomica Dominante Hendiduras palpebrales intensamente oblicuas, Disostosis mandibulofacial, coloboma, hipoplasia malar y fistulas ciegas. Ausencia de glándula parótida Inteligencia normal

70
CONCLUSION: El conocimiento de las patologías genéticas, nos guia hacia una practica pediátrica correcta. Las complicaciones de cada una de ellas, debe de ser estudiada en todo momento. Siempre hay que saber distinguir cuando un caso esta fuera de nuestro alcance y tener el valor de derivarlo a la especialidad correcta.

71
GRACIAS POR SU ATENCION AGRADECIMIENTOS: Dr. Hector Cuevas
Dra. Edna Aispuru

Presentaciones similares













>")





