Descargar la presentación
La descarga está en progreso. Por favor, espere

2
MECANISMOS DE INFLAMACION EN LA GOTA
Autores: Nathalie Busso, Alexander So. Objetivo del trabajo: Detallar los recientes avances en el entendimiento de los efectos de los cristales de MSU integrándolos con conocimientos previos referidos a la respuesta inflamatoria. CRISTALES DE URATO DE SODIO

3
¿QUE ES LA GOTA? Se produce por deposito de cristales de urato de sodio (MSU) en las articulaciones Los ataques agudos se caracterizan por dolor intenso e inflamación en la articulación Los síntomas iniciales son repentinos y violentos, teniendo lugar casi siempre de noche

4
CRISTALES DE URATO DE SODIO
Son la causa y no el efecto de la inflamación en la gota Son una señal de activación para la respuesta celular La RI innata es importante en la iniciación de la inflamación. Las cel. Del SI innato interaccionan con el tejido dañado y las cel muertas. Secretan citoquinas y quimiocinas que favorecen al proceso inflamatorio.

5
FACTORES DE LOS CRISTALES
CRISTALIZACIÓN DE URATO DE SODIO Ocurre cuando la concentración en plasma excede su solubilidad. Aunque la concentración puede no ser el único determinante. FACTORES MODIFICABLES QUE AFECTAN LA SOLUBILIDAD. Se cree que están en los fluidos biológicos ya que hay pacientes con hiperuricemia asintomáticos por largos periodos de tiempo. Además, pueden tener depósitos de MSU clínicamente inactivos, debido a una regulación adicional a nivel de la respuesta tisular. FACTORES PREDISPONENTES EN HIPERURICEMICOS Todavía son desconocidos los factores que llevan a la manifestación de gota. In vitro: Tº, pH, fuerza iónica, unión del urato a macromoléculas plasmáticas. Proteoglicanos incrementan la solubilidad del urato. (Katz y Shcubert).
.")
6
FACTORES DE LOS CRISTALES
Los cristales recubiertos con fragmentos de IgG son más inflamatorios. Apolipoproeína B desplaza a la IgG. Resolución de la artritis por gota aguda.

7
FACTORES DE LOS CRISTALES
Cristales de urato Son cuerpos extraños. Sobre las células Efectos TÓXICOS. El tamaño de los cristales No es un determinante mayor de su potencial inflamatorio. Variando el tamaño no afecta la producción de TNF por monocitos/macrof.

8
TIPOS DE CÉLULAS Las más estudiadas son neutrófilos y macrófagos.
Los neutrófilos son atraídos por factores quimiotácticos. Factores inducidos durante la inflamación de la gota son: IL-1beta, IL-8,CXCL1 y CFU-GM.

9
Los macrófagos no respondieron tan bien como los monocitos al MSU.
TIPOS DE CÉLULAS Los macrófagos no respondieron tan bien como los monocitos al MSU. La autorregulación de la inflamación por parte de los fagocitos puede explicar la naturaleza auto limitante de la gota. La diferencia de la respuesta celular parece estar ligada al estado del macrófago.(M1 y M2) En ratones con peritonitis: los macrófagos residentes juegan un papel importante en la iniciación de la respuesta tisular. Los monocitos también juegan un papel importante en la inflamación gotosa. Modelo air-pouch: con MSU se incremento la densidad de mastocitos y aumentó el contenido de Histamina en la burbuja. Son capaces de liberar IL-1 beta en la activación del inflamasoma NALP3 A través del LPS.
 En ratones con peritonitis: los macrófagos residentes juegan un papel importante en la iniciación de la respuesta tisular. Los monocitos también juegan un papel importante en la inflamación gotosa. Modelo air-pouch: con MSU se incremento la densidad de mastocitos y aumentó el contenido de Histamina en la burbuja. Son capaces de liberar IL-1 beta en la activación del inflamasoma NALP3. A través del LPS.")
10
IL-1 COMO MEDIADOR CRUCIAL EN LA INFLAMACIÓN DE LA GOTA.
Rol en el dolor y la inflamación de la gota. Bloqueo y deleción del IL-1R reducen la inflamación e hiperalgesia. IL-1RI y Myd88 son esenciales para la señal de transducción en el ensamblaje de IL-1.

11
COMPOSICIÓN DEL INFALAMASOMA NALP 3.ACTIVACIÓN POR MSU.

12
ROL DEL INFLAMASOMA NALP3
La IL-1beta es producida como una pro-molécula por macrófagos, monocitos, y células dendríticas Luego es clivada en una forma activa de la IL-1 beta para ser secretada. El clivaje es catalizado por la caspasa 1 .Requiere del inflamasoma para cumplir su función. Hay otras formas independiente de caspasa 1 que implican proteasas (neutrófilos y mastocitos) El inflamasoma es un complejo proteico citoplasmático. Compuesto por: proteína de la flia NLRP, proteína ASC y una caspasa inflamatoria. Para el inflamasoma NLRP3 es necesario un adaptador: proteína cardinal para reclutar una segunda caspasa 1, y así formar un dímero con la otra caspasa 1.
 El inflamasoma es un complejo proteico citoplasmático. Compuesto por: proteína de la flia NLRP, proteína ASC y una caspasa inflamatoria. Para el inflamasoma NLRP3 es necesario un adaptador: proteína cardinal para reclutar. una segunda caspasa 1, y así formar un dímero con la otra caspasa 1.")
13
ROL DEL INFLAMASOMA NALP3
Los macrófagos deficientes en componentes de este complejo fueron incapaces de secretar IL-1beta activa. La colchicina bloquea la maduración de IL-1 beta, por influencia en la endocitocis y/o presentación de los cristales al inflamasoma. Los cristales de MSU inician una cascada inflamatoria siendo el paso inicial la liberación de IL-1 beta activa.

14
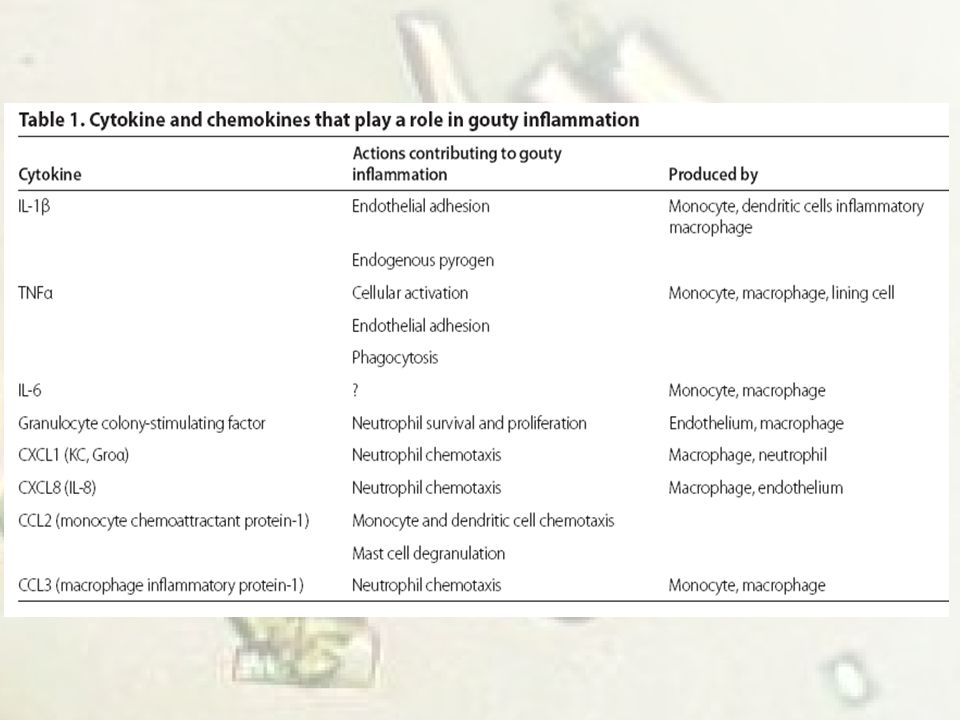
OTRAS CITOQUINAS IL-6 y TNF se incrementan cuando los monocitos entran en contacto con MSU. Datos sin publicar: bloqueo TNF no afecta el influjo de neutrófilos. Hay pacientes que respondieron a esta terapia. El rol de IL-6 es poco claro, es proinflamatoria. Quimiocinas juegan un papel importante en la inflamación: IL-8 y relacionadas(GRO )
")
16
TLR EN LA GOTA ¿Son importantes?
Son sensores y parte integral del SI innato. Ratones K.O TRL2 y 4 muestran disminución en la expresión de citoquinas inflamatorias y captación fagocítica. CD14 es compartida por los TLR 2 y 4 y es importante en la respuesta inflamatoria.(Ratones KO) La deficiencia de CD14 redujo la producción de IL-1beta sin efecto en la fagocitosis. En la etapa temprana de la RI innata es importante TREM-1(cel. Mieloides) que amplifica la respuesta.
 La deficiencia de CD14 redujo la producción de IL-1beta sin efecto en la fagocitosis. En la etapa temprana de la RI innata es importante TREM-1(cel. Mieloides) que amplifica la respuesta.")
17
COMPLEMENTO Y MEDIADORES SOLUBLES DE LA INFLAMACIÓN
MSU Activan de forma indirecta las cel. Inflamatorias. Activa la vía clásica del C ( no requiere Ig).Amplificada por IgG y Prot.CR. Se produce clivaje de C5 a C5a y C5b,por una convertasa de la superficie del cristal de MSU.
.Amplificada por IgG y Prot.CR. Se produce clivaje de C5 a C5a y C5b,por una convertasa de la superficie del cristal de MSU.")
18
CONCLUSIÓN Fagocitosis Activación del inflamasoma
Liberación de IL-1 beta Liberación de citoquinas proinflamatorias

19
Deficiencia de la HPRT: SINDROME DE LESCH-NYHAN

20
Metabolismo de las purinas

21
HERENCIA Recesiva ligada al X
Hombres : generalmente afectados. Mujeres: portadoras asintomaticas Codificada por un gen estructural en el brazo largo del cromosoma X en Xq26. Predominio: 1/ nacidos vivos en Canadá y 1/ en España.

22
Características clínicas
Sobreproducción de acido úrico. Manifestaciones neurológicas Trastornos hematológicos

23
Sobreproducción del ácido úrico
Síntomas renales y articulares asociados con la hiperuricemia. Presente en todos los pacientes con deficiencia de HPRT. Manifestaciones asociadas con la gota: artritis agudas, tofos, nefrolitiasis o urolitiasis, y enfermedad renal. Manifestaciones relacionadas con la hiperuricemia, incluyen: Cristales anaranjados en los pañales. Cristaluria: en los primeros años de vida. Artritis juvenil.

24
Síntomas neurológicos
TRASTORNO MOTOR Anormalidades motrices: espasticidad, coreoatetosis y balismo. Distonía de acción severa asociada a una insuficiencia completa de la HPRT. Disartria y disfagia, y opistótonos: se reportan con frecuencia. Signos del tracto corticoespinal, en años posteriores. Pacientes con insuficiencia parcial de la HPTR: dificultad en el habla, caminar distónico. DISCAPACIDAD COGNITIVA Pacientes con insuficiencia completa de HPTR: Retraso mental: leve a moderado. Déficit en la atención. Pacientes con insuficiencia parcial: Grado variable de retraso mental COMPORTAMIENTO COMPULSIVO AUTO-DESTRUCCTIVO Sólo en pacientes con defecto completo de la enzima. Auto-mutilación (2-16 años de edad). Comportamiento agresivo hacia otras personas.
. Comportamiento agresivo hacia otras personas.")
25
Aspectos hematológicos
Anemia megaloblática Anemia microcítica Hernia de hiato

26
Pacientes con insuficiencia de HPTR
CLASIFICACIÓN Pacientes con insuficiencia de HPTR Completa o con síndrome de Lesch-Nyhan Parcial o con síndrome de Kelly-Seegmiller

27
Otra clasificación incluye 3 grupos:
Lesch-Nyhan clásico o insuficiencia completa (LN) Insuficiencia de HPRT con manifestaciones neurológicas o hiperuricemia relacionada a la HPRT con discapacidad neurológica (HRND) Hiperuricemia relacionada a la HPRT (HRH) para pacientes sin manifestaciones neurológicas evidentes.
 Insuficiencia de HPRT con manifestaciones neurológicas o hiperuricemia relacionada a la HPRT con discapacidad neurológica (HRND) Hiperuricemia relacionada a la HPRT (HRH) para pacientes sin manifestaciones neurológicas evidentes.")
28
Cuatro grupos basados en información clínica, bioquimica, enzimática y molecular
Grupo 1: desarrollo normal sin síntomas neurológicos Grupo 2: síntomas neurológicos leves. Grupo 3: síntomas neurológicos severos Grupo 4: síndrome clásico de Lesch-Nyhan

29
DIAGNÓSTICO Clínico: La deficiencia de HPRT se caracteriza por:
Hiperuricemia con hiperuricosuria Manifestaciones neurológicas: distonía de acción severa, coreoatetosis, retardo mental de leve a moderado y auto-mutilación (en la forma completa)
")
30
POSIBILIDAD DE UNA DEFICIENCIA DE HPRT:
En el primer año de vida... La asociación de un retraso psicomotriz con hiperuricemia y/ o elevada relación del ácido úrico urinario/ creatinina. Un paciente con gota juvenil y elevada excreción de ácido úrico urinario.

31
Alta concentración de ácido úrico en suero
Bioquímico: Concentraciones plasmáticas medias de ácido úrico, hipoxantina y xantina, y sus tasas de excreción urinaria, son muy elevadas. Alta concentración de ácido úrico en suero Relación de ácido úrico urinario / creatinina como prueba de detección de enfermedades hereditarias del metabolismo de las purinas.

32
Aspectos bioquímicos:
ESTUDIOS POST MORTEM, revelaron una disfunción de neurotransmisores Disminución de la DOPAMINA. Aumento de serotonina y ácido 5-hidroindolacético LCR de pacientes con Lesch Nyhan: Disminución de ácido homovalínico, metabolito de DOPAMINA. Aumento en las concentraciones de hipoxantina y xantina.

33
Enzimático: Síndrome de Lesch-Nyhan Síndrome de Kelly-Seegmiller
Los pacientes presentan baja o indetectable actividad de HPRT en hemolizado, con aumento de la actividad adenina fosforribosiltransferasa (APRT). Actividad indetectable Síndrome de Lesch-Nyhan Actividad residual Síndrome de Kelly-Seegmiller
. Actividad indetectable. Síndrome de Lesch-Nyhan. Actividad residual. Síndrome de Kelly-Seegmiller.")
34
Molecular: HPRT: Codificada por un gen estructural que abarca 45 Kb en el brazo largo del cromosoma X en Xq26, y consta de nueve exones con una secuencia de codificación de 654 pb. Puede realizarse mediante la secuenciación de cDNA; en otros casos, puede ser necesaria la secuenciación del ADN genómico. Mutaciones puntuales: principal causa de deficiencia parcial de la enzima. Síndrome de Lesch-Nyhan es causado principalmente por mutaciones que modifican el tamaño de la enzima.

35
Diagnóstico diferencial
Retraso mental idiopático Parálisis cerebral Autismo Síndrome de Tourette Síndrome de Cornelia de Lange Alteraciones psiquiátricas graves

36
DIAGNÓSTICO PRENATAL Síndrome de Lesch-Nyhan
MUESTRAS: células amnióticas obtenidas por amniocentesis en las semanas de gestación, o células de vellosidades coriónicas obtenidos de las semanas de gestación. En ambos casos pueden llevarse a cabo un ensayo enzimático y el análisis molecular de la mutación en HPRT que causan la enfermedad .

37
TRATAMIENTO SOBREPRODUCCION DEL ACIDO URICO
Un inhibidor de xantina oxidasa: ALOPURINOL, que bloquea la conversión de xantina e hipoxantina en ácido úrico Reduce los niveles de urato sérico y ácido úrico en orina y por lo tanto evita que el ácido úrico produzca cristaluria, nefrolitiasis, artritis gotosa y tofos. Efecto adverso: litiasis xantina. SINDROME MOTOR La espasticidad y distonía puede ser manejado con BZD e inhibidores del ácido gamma-aminobutírico como el BACLOFENO. MANIFESTACIONES CONDUCTUALES Las BZD y CARBAMACEPINA son útiles para aminorar manifestaciones del comportamiento y la ansiedad.

38
Las causas de muerte son la neumonía y otras enfermedades infecciosas.
PRONÓSTICO Con el tratamiento adecuado con alopurinol, la función renal es por lo general conservada y los pacientes sobreviven hasta la segunda o tercera década de vida. Los pacientes con Lesch- Nyhan no pueden caminar y se limitan a una silla de ruedas. Con el tratamiento médico las conductas auto-mutilantes pueden ser manejadas apropiadamente. Las causas de muerte son la neumonía y otras enfermedades infecciosas.

Presentaciones similares


















>")


