Descargar la presentación
La descarga está en progreso. Por favor, espere

1
HEMOSTASIA Dr. Luis A. Mora B. Cátedra de Bioquímica UCIMED

3
HEMOSTASIA Definición:
Conjunto de mecanismos y procesos que mantienen la fluidez de la sangre y la integridad vascular.

4
Algunas características de la hemostasia
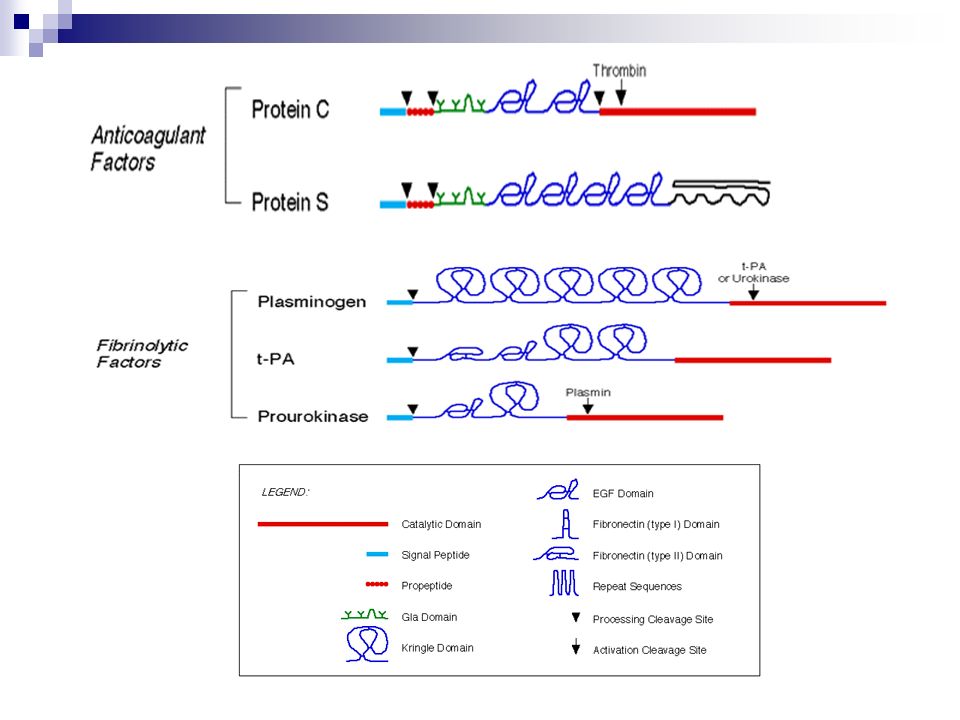
Propiedades: Mecanismo de defensa Inactivo en condiciones normales Constituido por: Proenzimas o zimógenos (proteasas) Procofactores Prot. reguladoras Características de las enzimas de la hemostasia: Actúan en la sangre Requieren regulación y control precisos Formadas por dominios catalíticos del tipo: COOH y NH2-terminales EGF Kringle Ácido carboxiglutámico y fibronectina
 Procofactores. Prot. reguladoras. Características de las enzimas de la hemostasia: Actúan en la sangre. Requieren regulación y control precisos. Formadas por dominios catalíticos del tipo: COOH y NH2-terminales. EGF. Kringle. Ácido carboxiglutámico y fibronectina.")
5
Algunas características de la hemostasia
Mecanismos: Contracción muscular Presión hística (tisular) Funciones celulares Funciones plasmáticas Factores indirectos: Sistemas enzimáticos: Complemento Quinina-calicreína Renina-angiotensina Factores reológicos Interacción intercelular
 Funciones celulares. Funciones plasmáticas. Factores indirectos: Sistemas enzimáticos: Complemento. Quinina-calicreína. Renina-angiotensina. Factores reológicos. Interacción intercelular.")
6
Fases de la hemostasia Fase vascular Fase plaquetaria Fase de los factores de la coagulación Fase del sistema fibrinolítico

7
Fase vascular Aportes del endotelio al subendotelio: Colágeno
Membrana basal Microfibrillas Mucopolisacáridos Elastinas y fibronectinas Aportes del endotelio a la sangre: Prostaciclina Factor tisular ( Factor III ) Factor von Willebrand ( FvW ) Antitrombina (AT) Trombomodulina Activador tisular del plasminógeno( tPA) Óxido nítrico y endotelinas.
 Factor von Willebrand ( FvW ) Antitrombina (AT) Trombomodulina. Activador tisular del plasminógeno( tPA) Óxido nítrico y endotelinas.")
8
Fase vascular Función del endotelio

9
SUPERFICIE PLAQUETARIA INACTIVA AL INHIBIDOR DEL tPA
Integración de las fases de la hemostasia CÉLULA ENDOTELIAL SUPERFICIE PLAQUETARIA PCa PS TROMBINA PC FV y FVIII INACTIVADOS INACTIVA AL INHIBIDOR DEL tPA TROMBOMODULINA Va VIIIa núcleo

10
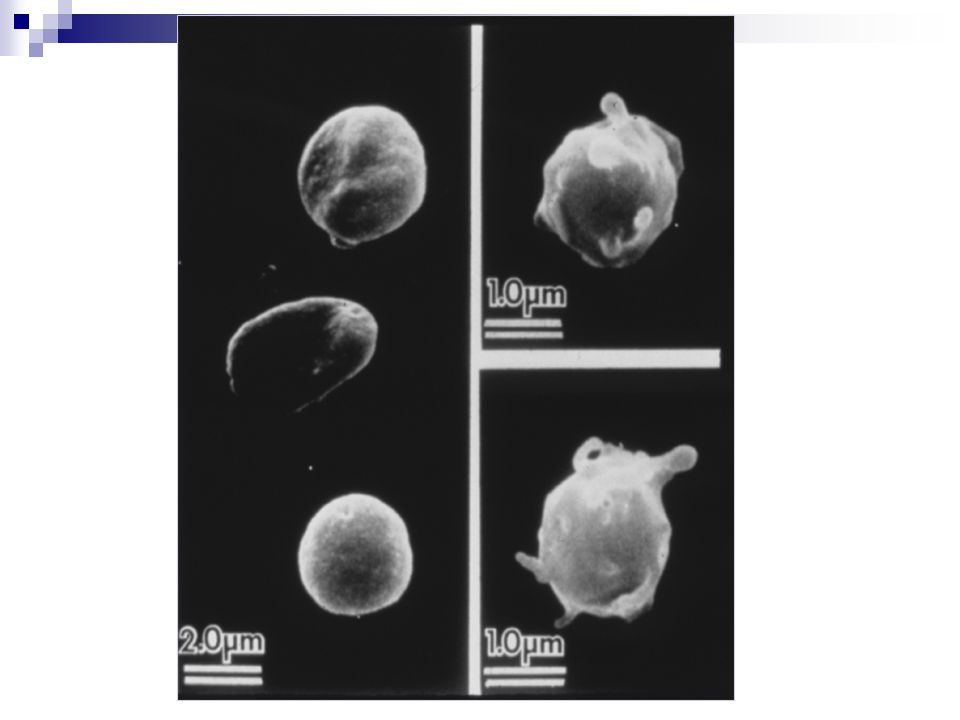
Fase plaquetaria Características de las plaquetas:
Unidad principal: Plaquetas o trombocitos discos redondos u ovalados. 2-4 micras de diámetro. normal en sangre périférica: /ul. se producen en médula ósea ( megacariocitos ) por estimulación de agentes humorales ( actividad de trombopoyetina): IL-6, IL 3 y GM-CSF. fragmentación citoplasmática. vida media: 8-12 días. Constituyentes de las plaquetas: Actina, miosina y trombostenina Calcio iónico Sistemas enzimáticos ATP y ADP prostaglandinas (tromboxano A2) Fase plaquetaria
 por estimulación de agentes humorales ( actividad de trombopoyetina): IL-6, IL 3 y GM-CSF. fragmentación citoplasmática. vida media: 8-12 días. Constituyentes de las plaquetas: Actina, miosina y trombostenina. Calcio iónico. Sistemas enzimáticos ATP y ADP. prostaglandinas (tromboxano A2) Fase plaquetaria.")
12
Fase plaquetaria Ultraestructura plaquetaria glucógeno glicocálix
Fosfolípidos plaquetarios Gránulos a específicos, factores de crecimiento, fibronectina, factor V, factor vW, fibrinógeno, b tromboglobulina, antagonistas de la heparina (PF4), trombospondina Gránulos densos: ADP, Ca2+, serotonina Sistema de túbulos densos Membrana plasmática glucógeno glicocálix Filamentos submembranosos (proteínas contráctiles) Sistema abierto de canales mitocondria Ultraestructura plaquetaria
, trombospondina. Gránulos densos: ADP, Ca2+, serotonina. Sistema de túbulos densos. Membrana plasmática. glucógeno. glicocálix. Filamentos submembranosos (proteínas contráctiles) Sistema abierto de canales. mitocondria. Ultraestructura plaquetaria.")
13
Fase plaquetaria Receptores glicoproteicos plaquetarios reaccionan con: Agentes agregantes Inhibidores Factores de la coagulación. Glicoproteína Ia : adhesión al colágeno. Glicoproteína Ib : unión de plaquetas al FvW (Síndrome de Bernard-Soulier) Glicoproteínas IIb/IIIa: unión de plaquetas al FvW (Trombastenia de Glanzman)
 Glicoproteínas IIb/IIIa: unión de plaquetas al FvW (Trombastenia de Glanzman)")
14
Fase plaquetaria Vaso sanguíneo GpIa Complejo GpIIb-IIIa Fibrinógeno
Colágeno GpIa Plaqueta Subendotelio Plaqueta Complejo GpIIb-IIIa Fibrinógeno Plaqueta Endotelio Plaqueta GpIb Factor de von Willebrand Colágeno Vaso sanguíneo

15
Fase plaquetaria Factor von Willebrand

16
Fase plaquetaria Características del Factor von Willebrand
Glicoproteína que forma complejo con Factor VIII Sintetizado en células endoteliales y megacariocitos PM 500 – kDA Control genético en cromosoma 12 Se une a receptores plaquetarios GPIb y GPIIb-IIIa Almacenamiento en : compartimento plaquetario ( gránulos alfa) y vascular ( cuerpos de Weibel-Palade ).
 y vascular ( cuerpos de Weibel-Palade ).")
17
Fase plaquetaria Agregación Fosfolípidos Plaqueta Ácido araquidónico
Fosfolipasa Tromboxano sintetasa Ácido araquidónico Endoperóxidos (PGG2 y PGH2) Tromboxano A2 Ciclo-oxigenasa (-) Aspirina (-) Sulfinpirazona ATP Prostaciclina (+) Célula endotelial Prostaciclina sintetasa AMPc (-) Dipiramidazole (-) AMP Ca+2 Agregación (+) Adenilato ciclasa (-) Fosfodiesterasa
 Tromboxano A2. Ciclo-oxigenasa. (-) Aspirina. (-) Sulfinpirazona. ATP. Prostaciclina (+) Célula endotelial. Prostaciclina sintetasa. AMPc (-) Dipiramidazole (-) AMP. Ca+2. Agregación. (+) Adenilato ciclasa. (-) Fosfodiesterasa.")
18
Fase de los factores de la coagulación
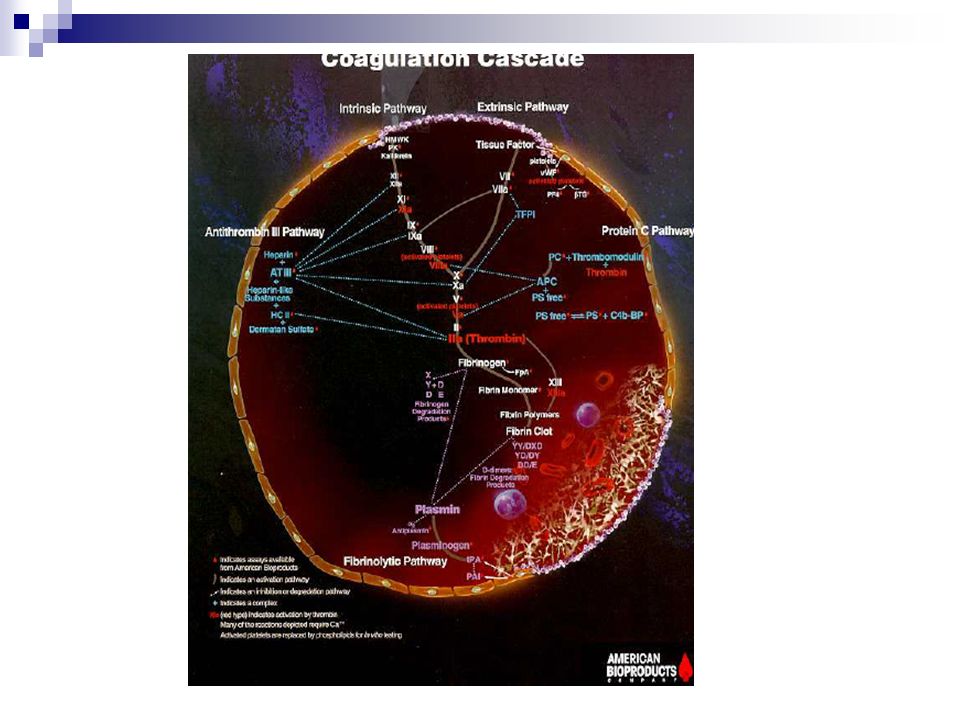
Cascada de la coagulación

19
Fase plaquetaria Etapas de la fase plaquetaria: 1) Adhesión
2) Agregación 3) Liberación 4) Retracción del coágulo
 Agregación. 3) Liberación. 4) Retracción del coágulo.")
21
Fase plaquetaria

22
Fase de los factores de la Coagulación

23
Fase de los factores de la coagulación
Etapas del mecanismo: Formación de tromboplastina Formación de trombina Formación de fibrina Lisis del coágulo de fibrina

24
Fase de los factores de la coagulación
Cascada de la coagulación

25
Fase de los factores de la coagulación

26
Fase de los factores de la coagulación

27
Factor II Protrombina * *o Cofactor II de Heparina.
Biosíntesis: Hígado, Vitamina K dependiente. PM: 70,000 daltons. Concentración Plasmática : 100 mg/L. Vida media In Vivo: 100 horas. Patología: Hipoprotrombinemia, autosómica recesiva. Ca++ Complejo Protrombinasa Fosfolípidos (Factor 3 plaquetario) Factor Va Factor Xa Fragmentos de activación Factor II Procoagulante Activación Factor IIa (Trombina) Anticoagulante Inhibición Factor IIa thrombina Anti- III * Anti- thrombina III Heparina Heparina *o Cofactor II de Heparina.
 Factor Va. Factor Xa. Fragmentos de activación. Factor. II. Procoagulante. Activación. Factor IIa. (Trombina) Anticoagulante. Inhibición. Factor. IIa. thrombina. Anti- III. * Anti- thrombina. III. Heparina. Heparina. *o Cofactor II de Heparina.")
28
Factor V Proacelerina, Factor Lábil Factor IIa Factor V Factor Va
Biosíntesis: Hígado, megacariocitos. PM: 330,000 daltons. Concentración en Plasma: 5-12 mg/L. Vida media In Vivo : 25 horas. Patología: Parahemofilia, autosómica recesiva. Factor IIa Procoagulante Factor V Activación Factor Va Inhibición Fragmentos Inactivos Anticoagulante Complejo Proteína C Proteína C Activada Ca++ Proteína S Fosfolípidos (Factor 3 plaquetario)
")
29
Factor VIII Factor Anti-hemofílico Factor IIa Factor Xa ó Factor VIII
Biosíntesis: Hígado, endotelio; Factor VIII Related Antigen, megacariocitos. PM (FVIII + vWF): million daltons Concentración Plasmática: 7 mg/L (vWF) Vida media In vivo: 10 horas (Factor VIII) Patología: Factor VIII-Hemofiia A, recesiva ligada al X. Enfermedad de von Willebrand, autosómica dominante. Factor IIa Factor Xa ó Procoagulante Factor VIII Activación Factor VIIIa Inhibición Fragmentos Inactivos Anticoagulant Complejo Proteína C Proteína C Activada Proteína S Fosfolípidos (Factor 3 plaquetario) Ca++
: million daltons. Concentración Plasmática: 7 mg/L (vWF) Vida media In vivo: 10 horas (Factor VIII) Patología: Factor VIII-Hemofiia A, recesiva ligada al X. Enfermedad de von Willebrand, autosómica dominante. Factor. IIa. Factor. Xa. ó. Procoagulante. Factor. VIII. Activación. Factor. VIIIa. Inhibición. Fragmentos. Inactivos. Anticoagulant. Complejo Proteína C. Proteína C Activada. Proteína. S. Fosfolípidos. (Factor 3 plaquetario) Ca++")
30
Fase de los factores de la coagulación

32
Fase de los factores de la coagulación
Clases funcionales Factores 1- Zimógenos II, VII, IX, X XI, XII 2- Cofactores III, V, VIII, KAPM 3- Fibrinógeno I 4- Inhibidores AT III, Prot. C, Prot. S 5- Factores de contacto XI, XII, Kalicreína Activación de los factores de contacto In vivo In vitro Colágeno vidrio Piel celita Estearato caolín Ácido úrico BaCo3 asbesto

33
Fase de los factores de la coagulación
Ubicación Factores 1- Suero y plasma VII, IX, X, XI, XII 2- Plasma (no en el suero) I, II, V, VIII Factores Vitamina K dependientes II, VII, IX, X, Prot. C, Prot. S No se adsorben con BaSO4 I, V, VIII, XI, XII Factores lábiles V, VIII
 I, II, V, VIII. Factores Vitamina K dependientes. II, VII, IX, X, Prot. C, Prot. S. No se adsorben con BaSO4. I, V, VIII, XI, XII. Factores lábiles. V, VIII.")
34
Fase de los factores de la coagulación
Precursores de los factores II, VII. IX, X, prot. C y prot. S (PIVKA) Ác. Glutámico (glu) Vitamina K Warfarina inhibe la reductasa Epóxido de Vitamina K Formas completas de los factores II, VII. IX, X, prot. C y prot. S Fosfolípidos plaquetarios se une a Ác. Glutámico gamma carboxilado (gla)
 Ác. Glutámico (glu) Vitamina K. Warfarina. inhibe. la reductasa. Epóxido de. Vitamina K. Formas completas de los factores II, VII. IX, X, prot. C y prot. S. Fosfolípidos. plaquetarios. se une a. Ác. Glutámico gamma carboxilado (gla)")
35
Fase de los factores de la coagulación
NH CH CH2 -OOC Gla CH COO- CO Ca+2 NH COO- Gla -OOC CH CH2 CH CO Parte externa de la membrana plaquetaria

36
Fase de los factores de la coagulación
Amplificación de la cascada enzimática: Iniciación Amplificación (ganancia por minuto) E0 Z E1 Z E2 Precursores Productos Ej. Fibrinógeno ej. fibrina 103 106 109
 E0. Z1 E1. Z2 E2. Precursores Productos. Ej. Fibrinógeno ej. fibrina")
37
Fase de los factores de la coagulación
Cascada de la coagulación

38
Fase de los factores de la coagulación
Acción de la trombina sobre el fibrinógeno

39
Fase de los factores de la coagulación
Polimerización de los monómeros de fibrina

40
Fase de los factores de la coagulación
Polimerización de los monómeros de fibrina

41
Fase de los factores de la coagulación
Acción de las proteasas de serina

42
Fase de los factores de la coagulación
Cascada de la coagulación

43
Cascada de la coagulación (mecanismos de retroalimentación)
")
44
Fase de los factores de la coagulación miento con cumarínicos
Pruebas relacionadas XII Vía intrínseca Vía extrínseca XI Factor tisular TTPa + IX TP VII Control de trata- miento con heparina VIII X Xa Va V Control de trata- miento con cumarínicos II Trombina II Fibrinógeno Fibrina XIII Fibrina estabilizada

45
Muchas gracias

46
Fase fibrinolítica

47
Estructura del fibrinógeno
Fase fibrinolítica Estructura del fibrinógeno

48
Fase fibrinolítica

49
Fase fibrinolítica

50
SUPERFICIE PLAQUETARIA INACTIVA AL INHIBIDOR DEL tPA
Integración de las fases de la hemostasia CÉLULA ENDOTELIAL SUPERFICIE PLAQUETARIA PCa PS TROMBINA PC FV y FVIII INACTIVADOS INACTIVA AL INHIBIDOR DEL tPA TROMBOMODULINA Va VIIIa núcleo

51
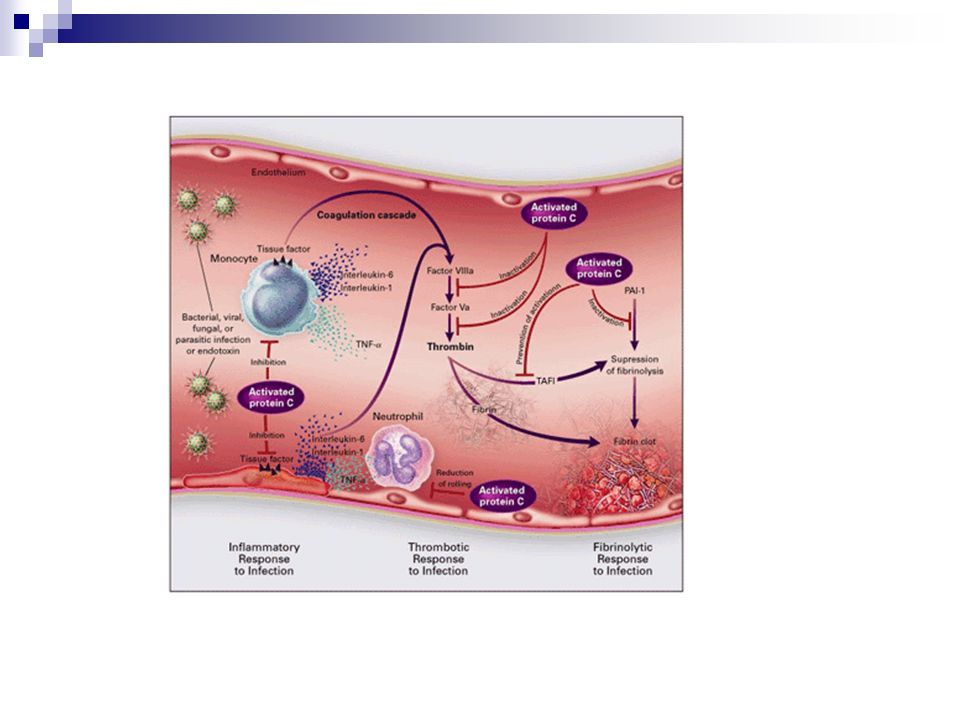
Bernard GR et al, NEJM 2001; 344: 699-709

52
Patologías asociadas a alteraciones de la coagulación
Defectos vasculares: Telangiectasia hemorrágica hereditaria (ovillos de capilares) Síndrome de Ehlers-Danlos (alteración del colágeno) Sangrado fácil Púrpura senil Púrpura asociada con infecciones Síndrome de Henoch-Schönlein (reac. de hipersensibilidad) Escorbuto Púrpura esteroidal Defectos plaquetarios: Cuantitativos: Trombocitopenia (PTI) Cualitativos: desorden en la función (EvW, Bernard-Soulier, Glanzman)
 Síndrome de Ehlers-Danlos (alteración del colágeno) Sangrado fácil. Púrpura senil. Púrpura asociada con infecciones. Síndrome de Henoch-Schönlein (reac. de hipersensibilidad) Escorbuto. Púrpura esteroidal. Defectos plaquetarios: Cuantitativos: Trombocitopenia (PTI) Cualitativos: desorden en la función (EvW, Bernard-Soulier, Glanzman)")
53
Patologías asociadas a alteraciones de la coagulación

54
Patologías asociadas a alteraciones de la coagulación
Hemofilia Factor deficiente Nombre Herencia VIII IX XI A B C Recesiva ligada al crom. X Autosómica dominante Hemofilia adquirida: Anticuerpos anti Factor VIII Terapia: crioprecipitados, concentrados de factores, DDAVP

55
xx xy xy xx xy xx Transmisión de la hemofilia 25 % mujer sana
Portadora Sana Sano Hemofílico xx xy xy xx 25 % mujer sana 25 % mujer portadora 25 % hombre sano 25 % hombre hemofílico 50 % mujer portadora 50 % hombre sano 25 % mujer hemofílica 25 % mujer portadora 25 % hombre sano 25 % hombre hemofílico xy xx

56
Patologías asociadas a alteraciones de la coagulación
Clínica de hemofilia Problemas hemorrágicos Por ligadura de cordón Por traumas Circuncisión Tipos de hemofilia Grave < 1% del factor Moderada >1 – 5 % del factor Leve >5 – 25 % del factor Hemorragias espontáneas Hemartrosis Artropatías Pocas hemorragias Hemartrosis y artropatías son poco frecuentes (raras) Traumáticas
 Traumáticas.")
57
Patologías asociadas a alteraciones de la coagulación
Desórdenes adquiridos de la coagulación Deficiencia de factores Vit. K dependientes Enf. hemorrágica del recién nacido Obstrucción biliar Mala absorción de Vit. K Tratamiento con antagonistas de Vit. K Enf. hepática Coagulación intravascular diseminada (CID) Inhibición de la coagulación Específicos (Anticuerpos anti FVIII) Inespecíficos (LES) Misceláneos (terapias, prod. de prot. M, sínd. transfusión masiva)
 Inhibición de la coagulación. Específicos (Anticuerpos anti FVIII) Inespecíficos (LES) Misceláneos (terapias, prod. de prot. M, sínd. transfusión masiva)")
58
Patologías asociadas a alteraciones de la coagulación
Causas de Coagulación Intravascular Diseminada (CID) Infecciones Septicemia por Gram neg., y Meningococcus sp Clostridium welchii, Malaria falciparum severa e inf. Viral. Malignidad Adenocarcinoma productor de mucina y LPA Complicaciones obstétricas Embolia del fluído amniótico, placenta previa, eclampsia Reacciones de hipersensibilidad Anafilaxis y transfusiones sanguíneas incompatibles Daño tisular extenso Por cirugía o trauma Misceláneos Fallo hepático, venenos de serpiente, quemaduras, hipotermia, hipoxia, malformaciones vasculares
 Infecciones. Septicemia por Gram neg., y Meningococcus sp. Clostridium welchii, Malaria falciparum severa e inf. Viral. Malignidad. Adenocarcinoma productor de mucina y LPA. Complicaciones obstétricas. Embolia del fluído amniótico, placenta previa, eclampsia. Reacciones de hipersensibilidad. Anafilaxis y transfusiones sanguíneas incompatibles. Daño tisular extenso. Por cirugía o trauma. Misceláneos. Fallo hepático, venenos de serpiente, quemaduras, hipotermia, hipoxia, malformaciones vasculares.")
59
Patologías asociadas a alteraciones de la coagulación
CID

60
Patologías asociadas a alteraciones de la coagulación
Trombo: masa sólida o tapón formado en la circulación a partir de componentes sanguíneos. Trombosis -Significado clínico: izquemia o embolización. -Arterial: reacción y acumulación plaquetaria por daño de la pared vascular. Ateroesclerosis de la pared arterial Ruptura de placa Activación plaquetaria Flujo sanguíneo reducido Pequeños émbolos de plaquetas y fibrina Deposición plaquetaria y formación de trombos

61
Patologías asociadas a alteraciones de la coagulación
-Venosa: generación de trombina en áreas de flujo sanguíneo enlentecido y sangre coagulada Liberac. de factor tisular Estasis No daño pared vascular Activación plaquetaria secundaria.

62
Patologías asociadas a alteraciones de la coagulación
Factores de riesgo en trombosis arterial (ateroesclerosis) Historia familiar positiva Sexo masculino Hiperlipidemia Hipertensión Diabetes mellitus Gota Policitemia Fumado Anormalidades del ECG Factor VII aumentado Fibrinógeno aumentado
 Historia familiar positiva. Sexo masculino. Hiperlipidemia. Hipertensión. Diabetes mellitus. Gota. Policitemia. Fumado. Anormalidades del ECG. Factor VII aumentado. Fibrinógeno aumentado.")
63
Patologías asociadas a alteraciones de la coagulación
Factores de riesgo en trombosis venosa: Relacionados con anormalidades en la coagulación Desórdenes hemostáticos hereditarios : (Requieren warfarina de por vida) Deficiencia de Antitrombina III Deficiencia de Proteína C Deficiencia de Proteína S Fibrinógeno anormal Plasminógeno anormal Desórdenes hemostáticos heredados o adquiridos: Niveles elevados de factor VII Niveles elevados de fibrinógeno Concentrados de factor IX Anticoagulante lúpico
 Deficiencia de Antitrombina III. Deficiencia de Proteína C. Deficiencia de Proteína S. Fibrinógeno anormal. Plasminógeno anormal. Desórdenes hemostáticos heredados o adquiridos: Niveles elevados de factor VII. Niveles elevados de fibrinógeno. Concentrados de factor IX. Anticoagulante lúpico.")
64
Patologías asociadas a alteraciones de la coagulación
Terapia con estrógenos Embarazo y puerperio. Cirugía ( abdominal y de cadera ) Trauma mayor Malignidad Infarto de miocardio Relacionados con estasis Fallo cardíaco Infarto Inmovilidad prolongada Obstrucción pélvica Síndrome nefrótico Deshidratación Hiperviscosidad Venas varicosas
 Trauma mayor. Malignidad. Infarto de miocardio. Relacionados con estasis. Fallo cardíaco. Infarto. Inmovilidad prolongada. Obstrucción pélvica. Síndrome nefrótico. Deshidratación. Hiperviscosidad. Venas varicosas.")
65
Patologías asociadas a alteraciones de la coagulación
Relacionados con factores desconocidos Edad Obesidad Sepsis HPN

66
Patologías asociadas a alteraciones de la coagulación
Terapia anticoagulante (heparina)
")
67
Patologías asociadas a alteraciones de la coagulación
Terapia anticoagulante (cumarínicos) Precursores de los factores II, VII. IX, X, prot. C y prot. S (PIVKA) Ác. Glutámico (glu) Vitamina K Warfarina inhibe la reductasa Epóxido de Vitamina K Formas completas de los factores II, VII. IX, X, prot. C y prot. S Fosfolípidos plaquetarios se une a Ác. Glutámico gamma carboxilado (gla)
 Precursores de los factores II, VII. IX, X, prot. C y prot. S (PIVKA) Ác. Glutámico (glu) Vitamina K. Warfarina. inhibe. la reductasa. Epóxido de. Vitamina K. Formas completas de los factores II, VII. IX, X, prot. C y prot. S. Fosfolípidos. plaquetarios. se une a. Ác. Glutámico gamma carboxilado (gla)")
68
Patologías asociadas a alteraciones de la coagulación

Presentaciones similares















 es una alteración fisiopatólogica sistémica, trombohemorrágica, que se presenta en algunas situaciones.>")



