Descargar la presentación
La descarga está en progreso. Por favor, espere

1
REGULACIÓN DE DISPOSITIVOS MÉDICOS
PHD BQ Nancy Fernández EE MS BME Antonio Glaría Bases de la electromedicina UNIVERSIDAD DE VALPARAÍSO Abril del 2005

2
DISPOSITIVO MEDICO ISO FIRST EDITION Cualquier instrumento, aparato, material u otros artículos, incluyendo software, usados solos o en combinación, definidos por el fabricante para ser usado en seres humanos solos o con el propósito de: diagnóstico, prevención, seguimiento, tratamiento o alivio de una enfermedad, daño o discapacidad; investigación, reemplazo o modificación de la anatomía o de un proceso fisiológico; regulación de la concepción. Y que no ejerza la acción principal que se desee obtener en el interior o en la superficie del cuerpo humano por medios farmacológicos, inmunológicos ni metabólicos, pero a cuya función puedan contribuir tales medios.

3
Fabricante y Distribuidores informan si un producto
REGULACIÓN DE DISPOSITIVOS MÉDICOS EN USA POR FDA (FOOD AND DRUG ADMINISTRATION) Medical Device Reports – MDRS: Fabricante y Distribuidores informan si un producto - Causa o contribuye a la muerte - Problemas de mal funcionamiento - Probabilidad que el evento vuelva a ocurrir.
 Medical Device Reports – MDRS: Fabricante y Distribuidores informan si un producto. - Causa o contribuye a la muerte. - Problemas de mal funcionamiento. - Probabilidad que el evento vuelva a ocurrir.")
4
REGULACIÓN DE DM COMUNIDAD EUROPEA
A inicios de 1980 solo Francia y Alemania regulaban algunos Reactivos de Diagnóstico derivados de la sangre Aparece el SIDA La industria comercializaba reactivos para VIH. Se cometieron errores (transfusiones bancos sangre). Los Estados Miembros percibieron la necesidad de regular productos para proteger la salud de la población: evitar infecciones por transmisión sexual y sanguínea . Se incluyó la rotulación en el etiquetado y otros requisitos para todos los Reactivos de Diagnóstico derivados de la sangre. La asociación de fabricantes europeos de PS-DIV solicitó la armonización de una Directiva para: Evitar reglamentaciones dispares entre Estados Rreducir costos de cumplimiento con el conjunto de tales reglamentaciones. Eliminar barreras al comercio. Permitir la libre circulación de productos. Explorar posibles acuerdos internacionales. Asegurar economías de escala en la producción
. Los Estados Miembros percibieron la necesidad de regular productos para proteger la salud de la población: evitar infecciones por transmisión sexual y sanguínea . Se incluyó la rotulación en el etiquetado y otros requisitos para todos los Reactivos de Diagnóstico derivados de la sangre. La asociación de fabricantes europeos de PS-DIV solicitó la armonización de una Directiva para: Evitar reglamentaciones dispares entre Estados. Rreducir costos de cumplimiento con el conjunto de tales. reglamentaciones. Eliminar barreras al comercio. Permitir la libre circulación de productos. Explorar posibles acuerdos internacionales. Asegurar economías de escala en la producción.")
5
REGULACION DE DISPOSITIVOS MÉDICOS EN CANADÁ - MEDICAL DEVICES BUREAU
La regulación Canadiense, data desde el año 1975 y establece un sistema obligatorio para la obtención de licencias, que se otorgan después de la exhaustiva verificación de todos los antecedentes presentados, para la comercialización en el mercado Canadiense

6
VIGILANCIA DE DISPOSITIVOS MÉDICOS
Esta área es realizada por Inspección de Productos, que efectua la vigilancia de Dispositivos Médicos en pre y post venta. Los problemas detectados determinan acciones por parte de esta área que pueden ir desde recomendaciones hasta cancelación de la licencia y/o retiro del producto del mercado Canadiense. Los problemas detectados en ocasiones son verificados y evaluados en una división especial

7
Regulaciones Sanitarias en Chile

8
Regulaciones Sanitarias en Chile
Fármacos, cosméticos y alimentos de uso médico, están controlados de acuerdo al Código Sanitario y D.S. 1876 Dispositivos Médicos, están controlados de acuerdo a la Ley y D.S. 825

9
DISPOSITIVOS MÉDICOS (M.D.)
ANTES DE 1997 1996 1995 1994 DISPOSITIVOS MÉDICOS (M.D.) Carencia de legislación que regule la calidad de estos productos. Carencia de normas nacionales acerca de especificaciones y características de calidad. Déficit de información acerca de procedimientos de fabricación. Se desconoce el impacto causado por productos en mal estado.
 Carencia de legislación que regule la calidad de estos productos. Carencia de normas nacionales acerca de especificaciones y características de calidad. Déficit de información acerca de procedimientos de fabricación. Se desconoce el impacto causado por productos en mal estado.")
10
(Medical Devices) Dispositivos Médicos
Cualquier instrumento, aparato, material u otros artículos, incluyendo software, usados solos o en combinación, definidos por el fabricante para ser usado en seres humanos solos o con el propósito de: diagnóstico, prevención, seguimiento, tratamiento o alivio de una enfermedad, daño o discapacidad; investigación, reemplazo o modificación de la anatomía o de un proceso fisiológico; regulación de la concepción. Dispositivos Médicos (Medical Devices) Y que no ejerza la acción principal que se desee obtener en el interior o en la superficie del cuerpo humano por medios farmacológicos, inmunológicos ni metabólicos, pero a cuya función puedan contribuir tales medios. ISO FIRST EDITION ISO First Edition
 Y que no ejerza la acción principal que se desee obtener en el interior o en la superficie del cuerpo humano por medios farmacológicos, inmunológicos ni metabólicos, pero a cuya función puedan contribuir tales medios. ISO FIRST EDITION ISO First Edition")
11
FASES PRINCIPALES DURANTE LA VIDA ÚTIL
DE LOS DISPOSITIVOS MÉDICOS (DE UN USO) CONCEPCIÓN Y DISEÑO FABRICACIÓN EMPAQUE Y ROTULADO ADVERTENCIAS VENTA USO DESECHO 1 2 3 4 5 6 7
 CONCEPCIÓN. Y. DISEÑO. FABRICACIÓN. EMPAQUE. Y. ROTULADO. ADVERTENCIAS. VENTA. USO. DESECHO")
12
PERSONAS QUE DIRECTAMENTE INTERVIENEN EN LAS
DIFERENTES FASES DE LOS D.M. (DE UN USO) CONCEPCIÓN Y DISEÑO FABRICACIÓN EMPAQUE Y ROTULADO ADVERTENCIAS VENTA USO DESECHO 1 2 3 4 5 6 7 FABRICANTE VENDEDOR USUARIO
 CONCEPCIÓN. Y. DISEÑO. FABRICACIÓN. EMPAQUE. Y. ROTULADO. ADVERTENCIAS. VENTA. USO. DESECHO FABRICANTE. VENDEDOR. USUARIO.")
13
RESPONSABILIDAD COMPARTIDA PARA LA SEGURIDAD Y EFICIENCIA DE D.M
Gobierno Público Fabricante Vendedor Usuario RESPONSABILIDAD COMPARTIDA COMUNICACIÓN PARTICIPACIÓN EDUCACIÓN ESTABLECER RIESGOS/COSTO

14
Marco Regulatorio para Dispositivos Médicos.
Ley No De acuerdo a Ley No de Marzo de 1997, el Instituto de Salud Pública de Chile será el organismo encargado de autorizar y fiscalizar a las entidades que realicen el referido control y certificación, debiendo a falta de organismos privados que desarrollen dichas tareas, ejecutarlas por si mismo. Las personas naturales o jurídicas que, a cualquier título, fabriquen, importen, comercialicen o distribuyan tales elementos, deberán realizar el respectivo control y certificación de su calidad en servicios, instituciones, laboratorios o establecimientos con autorización sanitaria expresa, otorgada de conformidad a lo dispuesto en el artículo 7° de este Código...

15
Decreto Supremo Nº 825 Artículo 22: Mediante decretos supremos fundados, dictados a través del Ministerio de Salud previo informe del Instituto, se hará efectiva progresivamente la aplicación de este reglamento a los distintos dispositivos y elementos médicos regulados por él, en los que deberá especificarse la clase a la que pertenecen y, por ende los controles regulatorios y requisitos aplicables a su respectiva verificación de la conformidad, según el caso. El informe del instituto se elaborará con la asesoría de una comisión de expertos, que propondrá la naturaleza y régimen aplicable al respectivo producto.

16
REGLAMENTO DE DM En agosto de 1999 se dicta el Reglamento N° 825 que hace operativa la Ley. El modelo utilizado en el Reglamento está basado en la regulaciones de USA, Canadá y la Comunidad Europea, adaptadas a realidad social y de salud de Chile y considerando que más del 90% de los D.M. que se comercializan son de importación. En la estrcutura básica de nuestra regulación se han considerado cinco elementos relevantes: 1.-Identificación del fabricante, distribuidor y producto previo a su comercialización. 2.-Exigencia a los fabricantes o distribuidores de contar con sistemas de calidad en sus procesos de fabricación. 3.-Clasificación de productos según riesgo (Clase I, II, III, IV) 4.-Evaluación de los productos, realizada por un organismo o empresa independiente, basada en normas nacionales o internacionales. 5.-Vigilancia durante la comercialización
 4.-Evaluación de los productos, realizada por un organismo o empresa independiente, basada en normas nacionales o internacionales. 5.-Vigilancia durante la comercialización.")
17
REGULACIÓN DE DM BENEFICIOS ESPERADOS
Incremento del Rol regulador del Estado sobre los productos utilizados en prestaciones de salud a la población. Disminución de recursos mal gastados por el Estado y los particulares en prestaciones de salud poco confiables. Asegurar a los usuarios: médicos, enfermeras, laboratorios y pacientes, la calidad de los dispositivos médicos. Garantía de la calidad de los productos que se comercializan, generados por el desarrollo tecnológico, estimulada por el control y la competencia entre los fabricantes.

18
Acceso a dispositivos médicos seguros y eficaces
REGULACIÓN DE DM OBJETIVOS ASEGURAR A LOS CHILENOS Acceso a dispositivos médicos seguros y eficaces Información sobre los riesgos y beneficios de los D.M. SISTEMA REGULATORIO QUE ASEGURE Evitar barreras injustificadas al acceso de dispositivos médicos Trato igualitario a la Industria nacional e internacional

19
¿CÓMO SE CLASIFICAN LOS DISPOSITIVOS MÉDICOS?
CLASIFICACIÓN DE DISPOSITIVOS MEDICOS DE ACUERDO AL NIVEL DE RIESGO ASOCIADO A SU USO Clase I: Incluye los dispositivos que presentan un bajo grado de riesgo. Clase II: Incluye los dispositivos que presentan un riesgo moderado. Clase III: Incluye los dispositivos que presentan un elevado potencial de riesgo. Clase IV: Incluye los dispositivos considerados los más críticos en materia de riesgos

20
¿CÓMO SE CLASIFICAN LOS DISPOSITIVOS MÉDICOS?
La clasificación de Artículos de Uso Médico, DM, se realizó recopilando información de clasificaciones de otros países tales como Canadá, USA y de la Comunidad Económica Europea. Los DM se han clasificado según riesgo y complejidad, de acuerdo a si son de uso in vitro; invasivos o no invasivos; activos o pasivos. En función de las características antes mencionadas hemos considerado la siguiente clasificación: CLASE I : Generalmente los DM no invasivos. Ej. gasa, tela adhesiva y termómetros, reactivos de diagnóstico in vitro (excepto aquellos usados en bancos de sangre) Los DM invasivos-pasivos. Ej. instrumental dental y quirúrgico. Los no invasivos in vitro incluyen la mayoría de los reactivos de diagnóstico.
 Los DM invasivos-pasivos. Ej. instrumental dental y quirúrgico. Los no invasivos in vitro incluyen la mayoría de los reactivos de diagnóstico.")
21
¿CÓMO SE CLASIFICAN LOS DISPOSITIVOS MÉDICOS?
CLASE II : Generalmente son: Los DM no invasivos-activos (usados para modificar composición de fluidos biológicos). Ej. equipos de infusión endovenosa. Los DM invasivos-activos. Ej. material de uso dental, amalgamas, material de ortodoncia. REACTIVOS DE DIAGNÓSTICO IN VITRO: se consideran en esta clase los reactivos de diagnóstico in vitro para autodiagnóstico (uso doméstico). CLASE III : Generalmente son: Los DM invasivos-activos, que se absorben o permanecen en el organismo por 30 días o más. Ej. suturas del tipo catgut, lentes intraoculares. Los no invasivos- activos ionizantes. Ej. DM que emiten radiaciones ionizantes, equipos radiológicos, equipos de radioterapia. REACTIVOS DE DIAGNÓSTICO IN VITRO: se consideran en esta clase los reactivos para VIH, hepatitis B y C, grupos sanguíneos etc.
. Ej. equipos de infusión endovenosa. Los DM invasivos-activos. Ej. material de uso dental, amalgamas, material de ortodoncia. REACTIVOS DE DIAGNÓSTICO IN VITRO: se consideran en esta clase los reactivos de diagnóstico in vitro para autodiagnóstico (uso doméstico). CLASE III : Generalmente son: Los DM invasivos-activos, que se absorben o permanecen en el organismo por 30 días o más. Ej. suturas del tipo catgut, lentes intraoculares. Los no invasivos- activos ionizantes. Ej. DM que emiten radiaciones ionizantes, equipos radiológicos, equipos de radioterapia. REACTIVOS DE DIAGNÓSTICO IN VITRO: se consideran en esta clase los reactivos para VIH, hepatitis B y C, grupos sanguíneos etc.")
22
¿CÓMO SE CLASIFICAN LOS DISPOSITIVOS MÉDICOS?
CLASE IV Generalmente son: Los DM invasivos-activos, aquellos usados para corregir defectos del sistema cardiovascular. Ej. marcapaso, válvulas cardíacas. Los invasivos no activos, que corrigen defectos del sistema nervioso. Ej. válvulas de descarga. DM DE TRANSICIÓN, AQUELLOS QUE REQUIEREN DE UN ANÁLISIS POR UN COMITÉ DE EXPERTOS PARA CLASIFICARLOS.

23
CLASE I CONTROLES REGULATORIOS REQUERIDOS SEGÚN CLASIFICACIÓN
PARA VERIFICACIÓN DE LA CONFORMIDAD CLASE I Identificación del producto, del fabricante y del distribuidor, especificación de sus características incluyendo rotulado del envase, instructivo interno y lote con número y código, y descripción de su funcionamiento. Declaración de materiales: lista de las partes y materias primas usadas en la fabricación, sus ensayos químicos y evaluación biológica si corresponde. Antecedentes de esterilización y almacenaje, si corresponde métodos de esterilización y controles respectivos; fecha de vencimiento y condiciones de almacenaje. Antecedentes nacionales o extranjeros que avalen la calidad del producto y/o de su producción. Evaluación del funcionamiento, si corresponde, según normas oficiales de la República de Chile y a falta de ellas por normas de organismos internacionales o estatales extranjeros especializados. Certificado para propósitos de exportación otorgado en el país de origen, autorizado por la autoridad estatal correspondiente y debidamente legalizado, en el caso dispositivos médicos que se importen a Chile.

24
CONTROLES REGULATORIOS REQUERIDOS SEGÚN CLASIFICACIÓN
PARA VERIFICACIÓN DE LA CONFORMIDAD CLASE III Todas las de la clase II. Literatura científica que respalde al producto. Estudios efectuados por el fabricante que demuestren la efectividad y seguridad del producto. Certificados de fabricación según sistema de calidad: modelo de aseguramiento de calidad en el diseño, desarrollo, producción, instalación y servicio, NCh-ISO o GMP. CLASE II Todas las de la clase I. Certificados de fabricación según sistema de calidad: modelo de aseguramiento de calidad en la producción, instalación y servicio, NCh-ISO o GMP.

25
PARA VERIFICACIÓN DE LA CONFORMIDAD
CONTROLES REGULATORIOS REQUERIDOS SEGÚN CLASIFICACIÓN PARA VERIFICACIÓN DE LA CONFORMIDAD CLASE IV Todas las de la clase III. Estudios realizados en grupos de pacientes representativos. Estudio de todos los riesgos inherentes al uso del dispositivo. Estudios biológicos realizados por el fabricante con relación al dispositivo.

26
ACTORES INVOLUCRADOS:
Instituto de Salud Pública de Chile Instituto Nacional de Normalización Organismos Certificadores Servicios de Salud

27
VERIFICACIÓN DE LA CONFORMIDAD DE D.M.
INSTITUTO DE SALUD PÚBLICA DE CHILE AUTORIZA INSTITUTO NACIONAL DE NORMALIZACIÓN ACREDITA ORGANISMOS CERTIFICADORES LABORATORIOS DE ENSAYO ACREDITADOS FABRICANTES PROVEEDORES DISPOSITIVO MÉDICO VERIFICACIÓN DE LA CONFORMIDAD DISPOSITIVO MÉDICO

28
DECRETO SUPREMO Nº 342 12 DE Mayo de 2004
REGULACIÓN DE DM DECRETO SUPREMO Nº 342 12 DE Mayo de 2004 1º.- Incorpórase al sistema de control que establece el artìculo 101 del Código Sanitario y sus reglamento, aprobado por decreto Nª 825 de 1998, del Minsterio de Salud, los productos y elementos de uso mèdico que se indican a continuaciòn. A.- Guantes Quirùrgicos de caucho para un uso Norma: 2173of91, ASTM 5712/99, Clase II. B.- Guantes de caucho para examen mèdico, Norma: NCh2181of91, ASTM 5712/99; Clase II. C.- Preservativos-Condones de làtex de caucho; Nch: Nch2224/1-2224/10of93; Clase III. Artículo transitorio: El presente decreto entrará en vigencia en el plazo de 270 dìas contandos desde su publicación en el Diario Oficial.

29
COMITÉ TÉCNICO NROAMILZIACIÓN DM
REGULACIÓN DE DM COMITÉ TÉCNICO NROAMILZIACIÓN DM 12 de Septiembre de 2006 Busca incorporar al sistema de control que establece el artìculo 101 del Código Sanitario y sus reglamento, aprobado por decreto Nª 825 de 1998, del Ministerio de Salud, mediante DS sucesivos, A.- 14 productos y elementos de uso médico que se indican. B.- Equipos electromédicos que deberán regirse de acuerdo con las NC2893/1(IEC ) y colaterales 1, 3 y 4 C.- Equipos electromédicos para electroenecefalografía de acuerdo con la NCh2893/226(IEC ) Busca incorporar Artículo transitorio: que establezca plazo de entradaen vigencia desde su publicación en el Diario Oficial.
 y colaterales 1, 3 y 4. C.- Equipos electromédicos para electroenecefalografía de acuerdo con la NCh2893/226(IEC ) Busca incorporar Artículo transitorio: que establezca plazo de entradaen vigencia desde su publicación en el Diario Oficial.")
30
SISTEMA DE VIGILANCIA DE DISPOSITIVOS MÉDICOS

31
OBJETIVO Mejorar la protección de la salud, tanto para pacientes como para usuarios, disminuyendo así la probabilidad de ocurrencia de incidentes adversos a través de una red de vigilancia: Con los Servicios de Salud públicos, privados, fabricantes y usuarios. Trabajo en conjunto para estandarizar los tiempos de notificación dependiendo de la gravedad del incidente. Proceso educativo constante

32
PROBLEMAS DE LOS DISPOSITIVOS MÉDICOS
Mal Funcionamiento: Mecánico o de software del dispositivo médico, defectos en la fabricación debido al diseño y problemas de los materiales utilizados. Problemas en el Uso: Rotulación incompleta del envase o del instructivo, instrucciones confusas o poco claras, problemas del diseño que dificultan el uso y adiestramiento deficiente del personal que los utiliza. Problemas Clínicos: Pacientes que tengan una condición deteriorada preexistente, alérgicos y/o sensibilizados al dispositivo médico, en individuos donde se utilizan productos contaminados que generan infecciones severas, etc.

33
CUÁNDO NOTIFICAR Muerte
Deterioro serio de la salud: Discapacidad permanente Deterioro serio condicional: Discapacidad reversible Mal funcionamiento de un DM que pudiera llegar a causar la muerte o el deterioro de la salud. Indicaciones erróneas, omisiones o insuficiencias en envase, instructivo o modo de empleo o manual de mantención del DM.

34
¿QUIÉNES NOTIFICAN EN UN SISTEMA DE VIGILANCIA?
Profesionales y personal de la salud, fabricantes o usuarios que consten o tomen conocimiento de incidentes o riesgos, relacionados con el mal funcionamiento, defectos de fabricación o reacciones adversas de los dispositivos médicos, antes, durante y después de ser utilizados en una prestación de salud.

35
¿CUÁL ES EL ROL DE LA AUTORIDAD SANITARIA?
Registrar y evaluar reportes y alertas informadas por los usuarios y empresas. Constante labor educativa de los actores involucrados. Implementar medidas correctivas que eviten la ocurrencia de nuevos eventos. Solicitar al fabricante y/o distribuidor estudios clínicos cuando sea necesario.

36
SISTEMA DE VIGILANCIA Descripción del Incidente
Detallada descripción de Causa del Incidente Justificación de la acción correctiva Acción posterior a la Investigación Aumento de la vigilancia post comercialización Acción correctiva y preventiva en relación al diseño y fabricación del dispositivo Retiro del mercado

37
VIGILANCIA DE LOS DISPOSITIVOS MÉDICOS, (D.M.)
Fecha * IDENTIFICACIÓN DEL USUARIO, OPERADOR DEL DISPOSITIVO Nombre: Profesión: Nombre Institución Rep.: Teléfono o fax: Servicio de Salud (si correponde): IDENTIFICACIÓN DEL DISPOSITIVO MÉDICO Nombre del Producto: Uso previsto: Nº de lote o serie: Método de esterilización si corresponde: Fecha de expiración si corresponde: IDENTIFICACIÓN DEL FABRICANTE, DISTRIBUIDOR O PROVEEDOR Nombre del Fabricante: Nombre del Distribuidor: Dirección: Teléfono / Fax: DATOS DEL INCIDENTE, FALLA, DEFECTO O ANOMALÍA Detección del incidente: Antes del uso Durante el uso Luego del uso Fecha, Hora: Descripción del procedimiento y del incidente ocurrido:
: IDENTIFICACIÓN DEL DISPOSITIVO MÉDICO. Nombre del Producto: Uso previsto: Nº de lote o serie: Método de esterilización si corresponde: Fecha de expiración si corresponde: IDENTIFICACIÓN DEL FABRICANTE, DISTRIBUIDOR O PROVEEDOR. Nombre del Fabricante: Nombre del Distribuidor: Dirección: Teléfono / Fax: DATOS DEL INCIDENTE, FALLA, DEFECTO O ANOMALÍA. Detección del incidente: Antes del uso. Durante el uso. Luego del uso. Fecha, Hora: Descripción del procedimiento y del incidente ocurrido:")
39
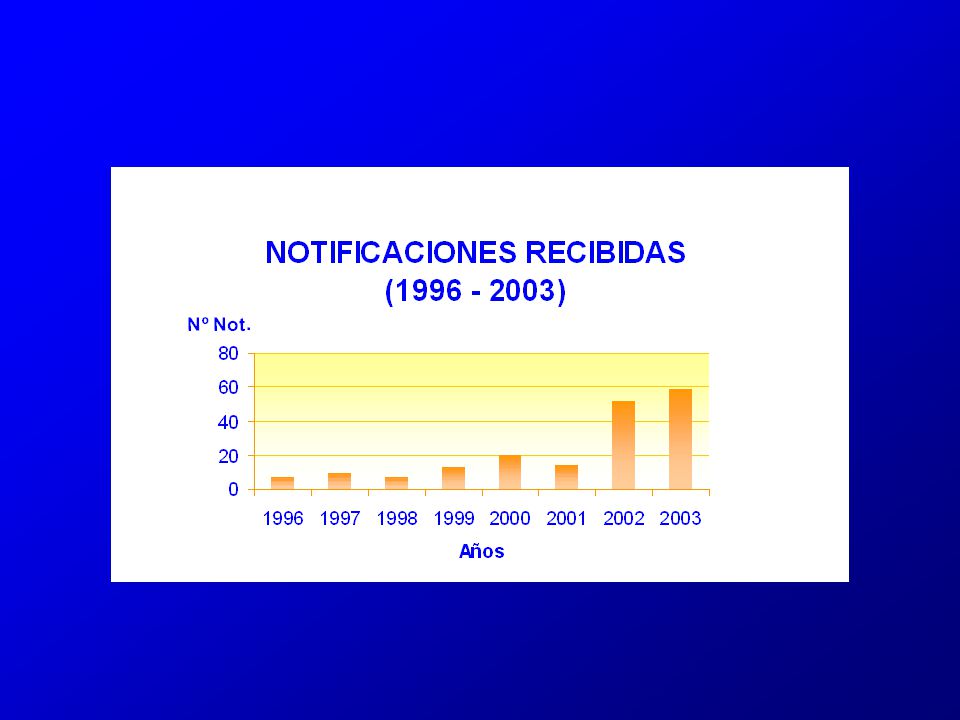
Notificaciones Recibidas:

40
Distribución Porcentual por Profesión (n=180)
Notificaciones Recibidas: Distribución Porcentual por Profesión

41
Pacientes, Profesionales, Técnicos
RED DE VIGILANCIA INSTITUTO DE SALUD PÚBLICA DE CHILE Evento Adverso 1 5 4 2 3 EVENTO ADVERSO Pacientes, Profesionales, Técnicos Fabricante Distribuidor Representante 1.-Reporte inicial 2.-Comunicación 3.-Reporte ISP 4.-Informe final 5.-Informe al Usuario

46
DISPOSITIVOS PARA LA SALUD ALERTAS
2006, Vol. 30 Nos. 27, 29, 30 DISPOSITIVOS PARA LA SALUD ALERTAS Una publicación del Sistema de Dispositivos para la Salud. Mejorando la seguridad y la calidad del cuidado del paciente mediante una gerencia tecnológica conocedora. En este mes recopilamos estas Alertas por la importancia que tienen para los países de América Latina y España.

47
*Parches de Vapor Novartis-Triaminic Posiblmente Pueden Causar Resultados Adversos a la Salud de ser Ingeridos por los Niños Accidentalmente. Almohadillas con Medicación [12-934] (Medicated Pads [12-934]) Dispositivo: Parches de Vapor (Consumibles) Identificación: Unidades distribuidas en Estados Unidos, Canadá, América Central y México. Fabricante: Novartis Consumer Health ], 200 Kimball Dr. Parsippany NJ Acción necesaria: Verifique que ha revisado la nota de prensa redactada por Novartis el 19 de Junio del 2006 (disponible en-línea en o el Public Health Advisory de la FDA (disponible en-línea en Identifique, separe, e inmediatamente descontinúe el uso de cualquier producto afectado en su inventario. Advierta a los pacientes que usan este producto en sus casas acerca de esta retirada del mercado. El fabricante comunica a los usuarios que tengan productos que están afectados que deben deshacerse de ellos o retornarlos a los puntos donde los compraron para que les devuelvan el dinero. Acción prioritaria: ALTA NOTA: Para más información usted puede suscribirse al Health Devices System para obtener acceso al sistema completo de las Alerta Médicas, este es el Acceso A7472 en la HDA del 7 de Julio del 2006 Vol.30 No.27, por
 Dispositivo: Parches de Vapor (Consumibles) Identificación: Unidades distribuidas en Estados Unidos, Canadá, América Central y México. Fabricante: Novartis Consumer Health ], 200 Kimball Dr. Parsippany NJ Acción necesaria: Verifique que ha revisado la nota de prensa redactada por Novartis el 19 de Junio del 2006 (disponible en-línea en o el Public Health Advisory de la FDA (disponible en-línea en Identifique, separe, e inmediatamente descontinúe el uso de cualquier producto afectado en su inventario. Advierta a los pacientes que usan este producto en sus casas acerca de esta retirada del mercado. El fabricante comunica a los usuarios que tengan productos que están afectados que deben deshacerse de ellos o retornarlos a los puntos donde los compraron para que les devuelvan el dinero. Acción prioritaria: ALTA. NOTA: Para más información usted puede suscribirse al Health Devices System para obtener acceso al sistema completo de las Alerta Médicas, este es el Acceso A7472 en la HDA del 7 de Julio del 2006 Vol.30 No.27, por")
48
Extracto del HDA 2006, Vol.30 No.29 Agujas de Biopsia Coaxiales tipo TruGuid fabricadas por la Bard: Es Posible que El Estilete Interior sea Más Largo de lo Supuesto Agujas, para Biopsia [12-734] (Needles, Biopsy [12-734]) Identificación: Catálogo No. C1816B; Listado de Dispositivos No. E148687; Lote No. REP JO196: unidades distribuidas en California, Florida, Montana, New Jersey, Texas, Canadá y América Latina. Fabricante: Bard Peripheral Vascular Div. C R Bard Inc. [427969], PO Box 1740, Tempe AZ Problema: Cuando el estilete interior de las agujas de biopsia es insertado como un componente dentro de la cánula la porción expuesta del estilete es posible que sea hasta 8mm más larga de lo esperado. Este problema es el resultado de un cambio en el proceso de fabricación que introdujo una variabilidad en el largo del estilete interior. El fabricante inicio una revocación mediante una carta fechada el 1ro. de Mayo del La FDA ha denominado esta revocación “Class II Recall No. Z ”. Health Canadá ha designado esta revocación “Type II Recall No Acción necesaria: Verifique que ha recibido la carta fechada el 1ro. de Mayo del 2006 y la “Recall and Effectiveness Check Form” enviada por la Bard. Identifique y separe cualquier producto afectado en su inventario. Aunque no tenga ningún producto afectado complete la forma “Recall and Effectiveness Check Form” y devuélvala a la Bard por fax al (fuera de los Estados Unidos). Para más información póngase en contacto con la Bard al teléfono indicado. Acción prioritaria: ALTA NOTA: Para más información usted puede suscribirse al Health Devices System para obtener acceso al sistema completo de las Alerta Médicas, este es el Acceso A7409 en la HDA del 21 de Julio del 2006 Vol.30 No.29, por
 Identificación: Catálogo No. C1816B; Listado de Dispositivos No. E148687; Lote No. REP JO196: unidades distribuidas en California, Florida, Montana, New Jersey, Texas, Canadá y América Latina. Fabricante: Bard Peripheral Vascular Div. C R Bard Inc. [427969], PO Box 1740, Tempe AZ Problema: Cuando el estilete interior de las agujas de biopsia es insertado como un componente dentro de la cánula la porción expuesta del estilete es posible que sea hasta 8mm más larga de lo esperado. Este problema es el resultado de un cambio en el proceso de fabricación que introdujo una variabilidad en el largo del estilete interior. El fabricante inicio una revocación mediante una carta fechada el 1ro. de Mayo del La FDA ha denominado esta revocación Class II Recall No. Z Health Canadá ha designado esta revocación Type II Recall No Acción necesaria: Verifique que ha recibido la carta fechada el 1ro. de Mayo del 2006 y la Recall and Effectiveness Check Form enviada por la Bard. Identifique y separe cualquier producto afectado en su inventario. Aunque no tenga ningún producto afectado complete la forma Recall and Effectiveness Check Form y devuélvala a la Bard por fax al (fuera de los Estados Unidos). Para más información póngase en contacto con la Bard al teléfono indicado. Acción prioritaria: ALTA. NOTA: Para más información usted puede suscribirse al Health Devices System para obtener acceso al sistema completo de las Alerta Médicas, este es el Acceso A7409 en la HDA del 21 de Julio del 2006 Vol.30 No.29, por")
49
Extracto del HDA 2006, Vol.30 No.30 Posibilidad que El Dispositivo Neuroestimulador Implantable de la Medtronic-Kinetra, falle. Estimuladores, Eléctricos, del Cerebro, contra Temblores [18-468] Stimulators, Electrical, Brain, Tremor [18-468] Identificación: Códigos del Producto NFD621879S, NFD621891S, NFD621901S, NFD621976S, NFD621981S, NFD622170S, NFD622185S, NFD622189S, NFD622201S, NFD622247S, NFD622279S, NFD622345S, NFD622349S, NFD622372S, NFD622382S, NFD622387S, NFD622400S, NFD622414S, NFD622466S, NFD622542S, NFD622564S, NFD622629S, NFD622664S, NFD623035S; 24 unidades distribuidas en Estados Unidos, Bélgica, Francia, Alemania, Italia, España, Suiza y en Gran Bretaña. Distribuidor: Medtronic Inc Neurological Div [105368], 710 Medtronic Pkwy, Min. MN Fabricante: Medtronic Inc [101809], Rd 31 Km 24 Hm4, Ceiba Norte Industrial Park, Juncos PR 00777 Problema: Es posible que los dispositivos anteriormente señalados fallen debido a interrupciones en los alambres de unión entre el circuito híbrido y la batería. El fallo del dispositivo provocará el retorno de las condiciones médicas existentes con anterioridad en el paciente. Acción necesaria: Verifique que ha recibido la carta “Urgent Device Recall important Patient Management” de la Medtronic. Identifique si tiene alguno de los productos afectados en su inventario. Para mayor información, póngase en contacto con la Medtronic por el teléfono Fuente: En los Estados Unidos “Food and Drug Administration (FDA), Center for Devices and Radiological Health”. Medical Device recalls: Class 3 recall - Kinetra implantable neuroestimulador [en-línea] Disponible: FDA Enforcement Rep 2006 Jun7. Acción Prioritaria: ALTA NOTA: Para más información usted puede suscribirse al Health Devices System para obtener acceso al sistema completo de las Alerta Médicas, este es el Acceso A7401 en la HDA del 28 de Julio del 2006 Vol.30 No. 30, por
, Center for Devices and Radiological Health . Medical Device recalls: Class 3 recall - Kinetra implantable neuroestimulador [en-línea] Disponible: ID= FDA Enforcement Rep 2006 Jun7. Acción Prioritaria: ALTA. NOTA: Para más información usted puede suscribirse al Health Devices System para obtener acceso al sistema completo de las Alerta Médicas, este es el Acceso A7401 en la HDA del 28 de Julio del 2006 Vol.30 No. 30, por")
50
ECRI AGENCIA NO LUCRATIVA Renuncia: La información emitida por este servicio es para su referencia solamente y no constituye una opinión o consejo oficial por parte de la ECRI. Los enlaces para sitios de la internet "websites" fuera del son para su comodidad. La ECRI no es responsable del contenido o la información que se encuentra en otros sitios de la internet y no realiza control editorial o de otra forma sobre aquellos sitios. Arq. Amalia V. Patino Programas Internacionales ECRI 5200 Butler Pike Plymouth Meeting, PA U.S.A. Tel: X 5190 Fax: Copyright 2006 ECRI

Presentaciones similares