Descargar la presentación
La descarga está en progreso. Por favor, espere

1
Patologías de la membrana plasmática
Objetivo: Conocer acerca de algunas patologías asociadas a la disfunción de alguno de los componentes de la membrana celular.

2
PATOLOGÍAS DE PROTEÍNA DE TRANSPORTE

3
Síndrome de Fanconi

4
El síndrome de Fanconi es un defecto generalizado del transporte de aminoácidos, glucosa, fosfato, ácido úrico, sodio, potasio, bicarbonato y proteínas a través del túbulo proximal. El síndrome de Fanconi idiopático2 se puede heredar como: Rasgo autosómico dominante Autosómico recesivo Ligado al cromosoma X.

5
causas El síndrome de Fanconi puede ser causado por genes defectuosos o puede aparecer posteriormente en la vida debido a daño renal. Algunas veces, se desconoce su causa. En los niños, las causas comunes de este síndrome son defectos genéticos que afectan la capacidad del cuerpo para descomponer ciertos compuestos, como: Cistina (cistinosis) Fructosa (intolerancia a la fructosa) Galactosa (galactosemia) Glucógeno (enfermedades por almacenamiento de glucógeno) Otras causas en niños comprenden: Exposición a metales pesados como el plomo, el mercurio y el cadmio Enfermedad de Lowe, un raro trastorno genético de los ojos, el cerebro y los riñones Enfermedad de Wilson
 Fructosa (intolerancia a la fructosa) Galactosa (galactosemia) Glucógeno (enfermedades por almacenamiento de glucógeno) Otras causas en niños comprenden: Exposición a metales pesados como el plomo, el mercurio y el cadmio. Enfermedad de Lowe, un raro trastorno genético de los ojos, el cerebro y los riñones. Enfermedad de Wilson.")
6
En los adultos, el síndrome de Fanconi puede ser causado por diversas cosas que provocan daño a los riñones, como: Ciertos medicamentos como azatioprina, cidofovir, gentamicina y tetraciclina Trasplante de riñón Enfermedad por precipitación de las cadenas ligeras Mieloma múltiple Amiloidosis primaria SINTOMAS Eliminar grandes cantidades de orina, lo cual puede llevar a deshidratación Dolor en los huesos Debilidad

7
Tratamiento Cuando la afectación incluye los huesos puede tratarse con fosfatos y suplementos de vitamina D que se administrarán por vía oral. Si la enfermedad evoluciona hacia una insuficiencia renal puede ser necesario un trasplante de riñón. En los casos de acidosis sanguínea elevada se puede contrarrestar el problema bebiendo bicarbonato sódico. En cualquier caso, a día de hoy, hay que decir que el síndrome de Fanconi no tiene cura.

8
PATOLOGÍAS DE PROTEÍNAS RECEPTORAS

9
Acondroplasia

10
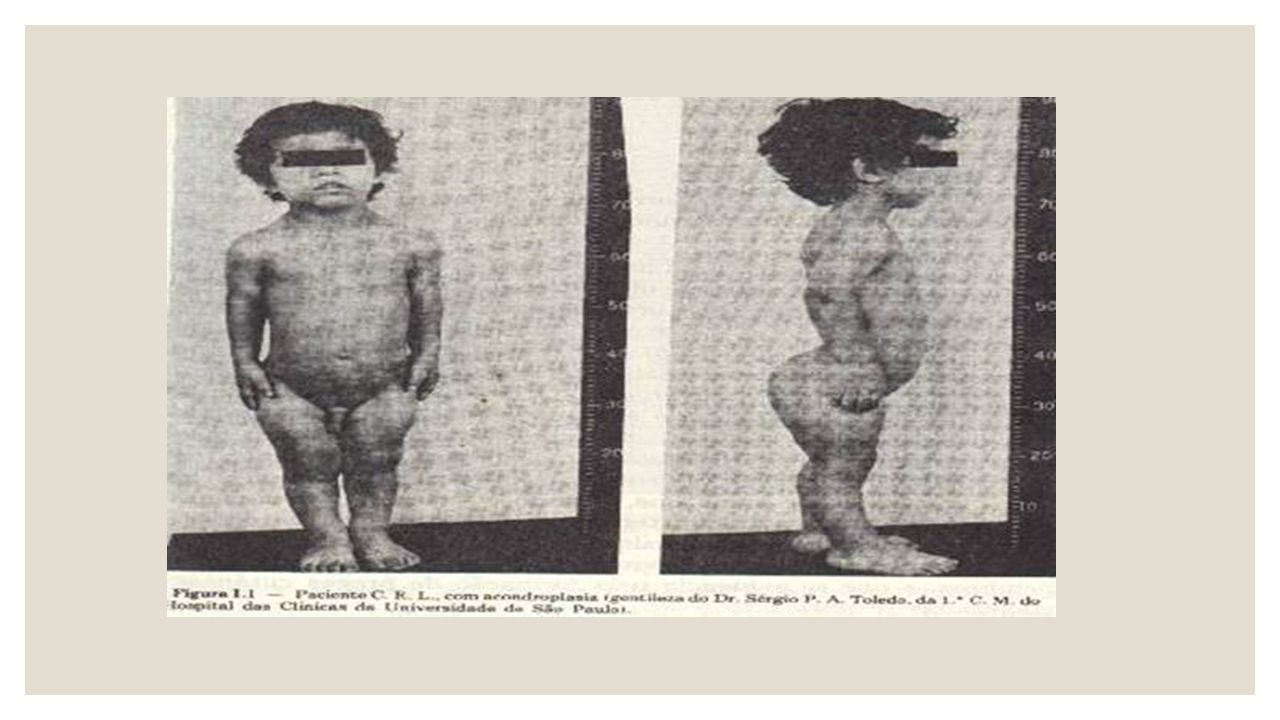
ACONDROPLASIA La acondroplasia es un trastorno genético que causa enanismo (estatura corta). Es un trastorno en el cual los huesos y cartílagos no crecen normalmente. Es la causa más común de enanismo. Los pacientes que padecen esta condición alcanzan una estatura de crecimiento total menor que 1,2 metros. El mayor acortamiento ocurre en el húmero (el hueso entre el hombro y el codo) y el fémur (el hueso entre la cadera y la rodilla). También puede haber sub-desarrollo en el rostro. La acondroplasia es la forma más común de estatura corta desproporcional hereditaria. Ocurre de 1 en cada 26,000 a 1 en cada 40,000 nacimientos vivo
. Es un trastorno en el cual los huesos y cartílagos no crecen normalmente. Es la causa más común de enanismo. Los pacientes que padecen esta condición alcanzan una estatura de crecimiento total menor que 1,2 metros. El mayor acortamiento ocurre en el húmero (el hueso entre el hombro y el codo) y el fémur (el hueso entre la cadera y la rodilla). También puede haber sub-desarrollo en el rostro. La acondroplasia es la forma más común de estatura corta desproporcional hereditaria. Ocurre de 1 en cada 26,000 a 1 en cada 40,000 nacimientos vivo.")
12
La acondroplasia es un trastorno genético
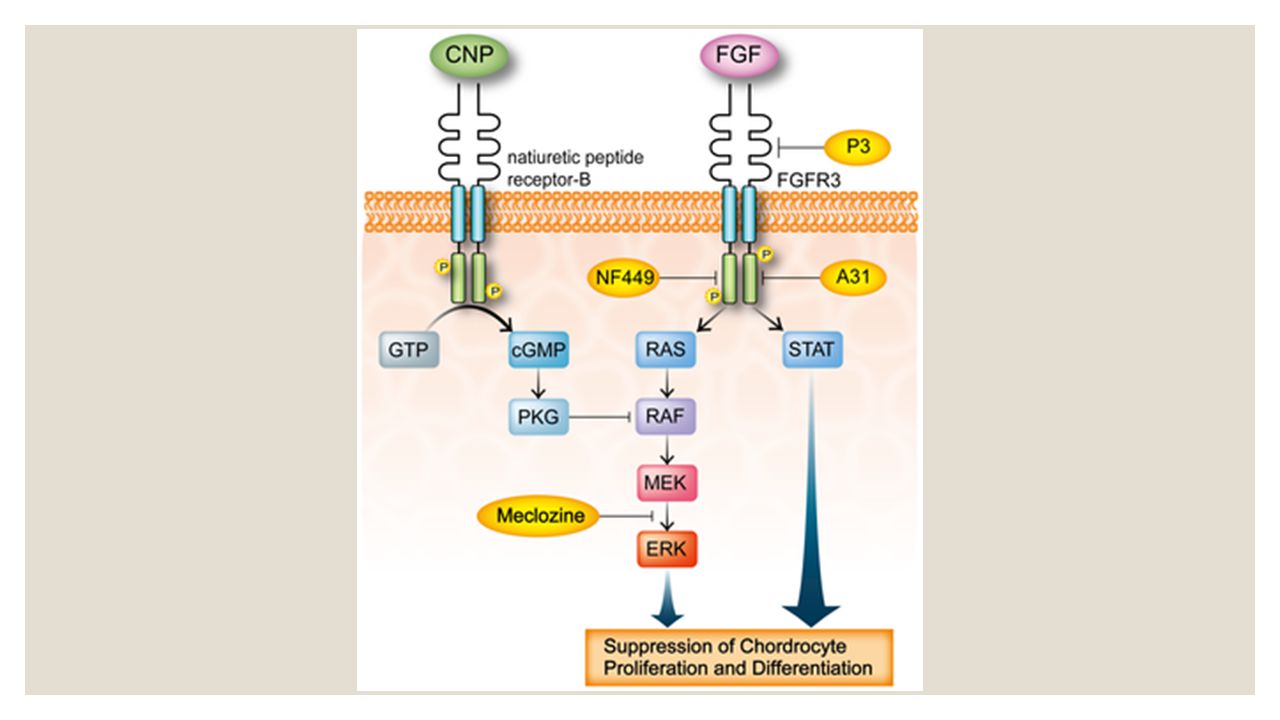
La acondroplasia es un trastorno genético. Es causado por mutaciones en el gen FGFR3. Este gen evita el crecimiento de cartílago en la placa de crecimiento. El FGFR3 codifica una proteína llamada receptor 3 del factor de crecimiento de fibroblastos. Esta proteína es el lugar de acción de un factor de crecimiento principal responsable del alargamiento de los huesos. Cuando este factor de crecimiento no puede actuar correctamente por la ausencia de su receptor, el crecimiento de los huesos, en el cartílago de la placa de crecimiento, se hace más lento. Esto conlleva a huesos más cortos, huesos en forma anormal, y estatura más corta.

14
Hipercolestemia familiar

15
Hipercolesterolemia familiar La hipercolesterolemia familiar (HF) es una enfermedad hereditaria que se expresa desde el nacimiento, y que cursa con un aumento en las concentraciones plasmáticas de colesterol, principalmente del colesterol transportado por las lipoproteínas de baja densidad (c-LDL) Es un trastorno hereditario que provoca niveles de colesterol LDL ("malo") muy altos. La afección empieza al nacer y puede causar ataques cardíacos a temprana edad.

17
La hipercolesterolemia familiar es un trastorno genético causado por un defecto en el cromosoma 19.
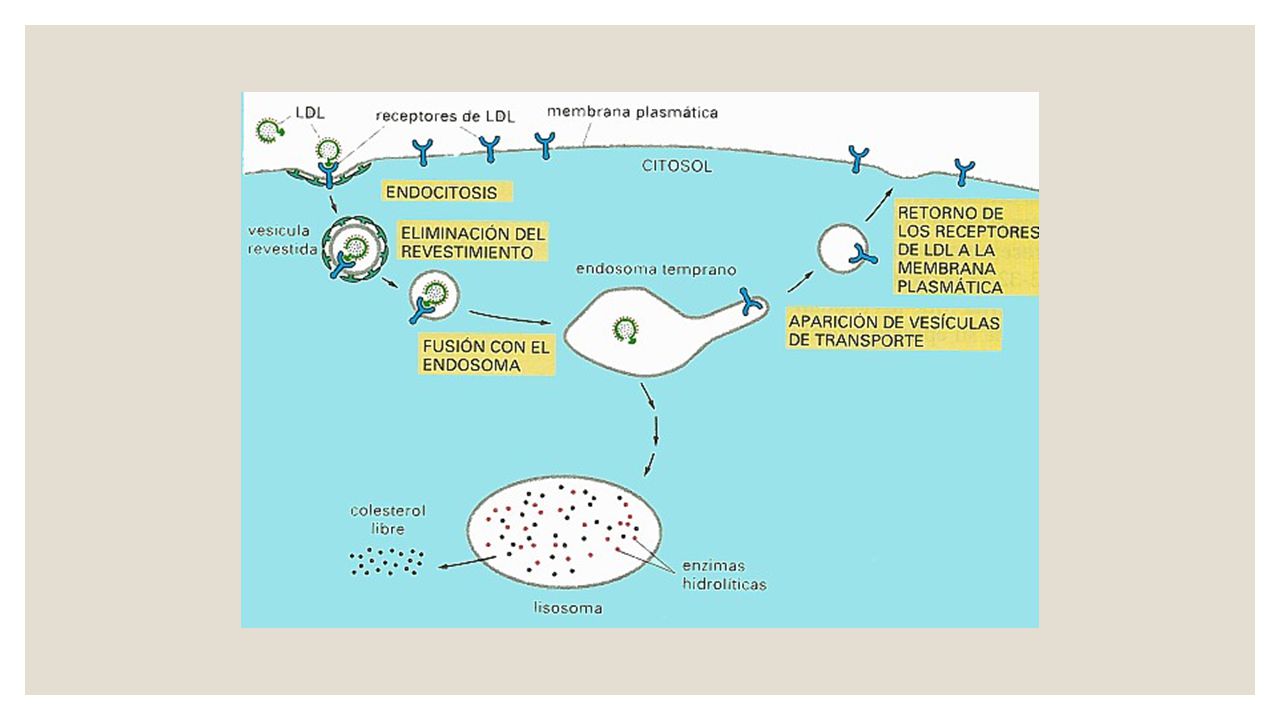
El defecto hace que el cuerpo sea incapaz de eliminar la lipoproteína de baja densidad (colesterol LDL o "malo") de la sangre. Esto provoca niveles altos de colesterol LDL en la sangre, lo cual hace que uno sea más propenso a presentar estrechamiento de las arterias a raíz de ateroesclerosis a temprana edad. La afección se hereda típicamente de forma autosómica dominante, lo cual significa que sólo se necesita recibir un gen anormal de uno de los padres con el fin de heredar la enfermedad. En casos excepcionales, un niño puede heredar el gen de ambos padres. Cuando esto ocurre, el incremento en los niveles de colesterol es mucho más grave. El riesgo de cardiopatía y ataques cardíacos es más alto incluso en la niñez.
 de la sangre. Esto provoca niveles altos de colesterol LDL en la sangre, lo cual hace que uno sea más propenso a presentar estrechamiento de las arterias a raíz de ateroesclerosis a temprana edad. La afección se hereda típicamente de forma autosómica dominante, lo cual significa que sólo se necesita recibir un gen anormal de uno de los padres con el fin de heredar la enfermedad. En casos excepcionales, un niño puede heredar el gen de ambos padres. Cuando esto ocurre, el incremento en los niveles de colesterol es mucho más grave. El riesgo de cardiopatía y ataques cardíacos es más alto incluso en la niñez.")
20
PATOLOGÍAS DE PROTEÍNAS DE CONTACTO

22
El cáncer: es un crecimiento tisular producido por la proliferación continua de células anormales con capacidad de invasión y destrucción de otros tejidos. El cáncer, que puede originarse a partir de cualquier tipo de célula en cualquier tejido corporal, no es una enfermedad única, sino un conjunto de enfermedades que se clasifican en función del tejido y de la célula de origen. Origen del cáncer Entre dichos factores se encuentran La herencia, Los productos químicos Las radiaciones ionizantes Las infecciones o virus Los traumas. El cáncer es un proceso genético. Las alteraciones genéticas pueden ser heredadas o producidas en alguna célula por un virus o por una lesión provocada de manera externa

23
Tres principales subtipos
los sarcomas, que proceden del tejido conectivo como huesos, cartílagos, nervios, vasos sanguíneos, músculos y tejido adiposo. Los carcinomas, que proceden de tejidos epiteliales como la piel o los epitelios que tapizan las cavidades y órganos corporales, y de los tejidos glandulares de la mama y de la próstata. Las leucemias y los linfomas, que incluyen los cánceres de los tejidos formadores de las células sanguíneas.

24
Herencia: se calcula que de un 5 a un 10% de los cánceres tienen un origen hereditario. El cáncer de mama y el colon es un ejemplo de ello. Una forma de retinoblastoma sólo aparece cuando está ausente un gen específico. Estos genes, denominados genes supresores tumorales o antioncogenes, previenen en condiciones normales la replicación celular, su ausencia elimina el control normal de la multiplicación celular. En algunos trastornos hereditarios, los cromosomas tienen una fragilidad intrínseca; estos procesos conllevan un riesgo elevado de cáncer.

25
Sustancias químicas El alquitrán de hulla y sus derivados se consideran altamente cancerígenos. El cigarrillo es otro agente cancerígeno; se ha determinado que la muerte por cáncer de pulmón es 6 veces mayor entre fumadores que entre no fumadores. El alcohol es también un importante promotor; su abuso crónico incrementa de manera importante el riesgo de cánceres que son inducidos por otros agentes. Se ha encontrado que, en países donde la contaminación de alimentos por mohos es frecuente, la incidencia de cáncer de hígado y de estómago es alta. El arsénico se asocia con cáncer del pulmón.

26
Radiaciones La radiación produce cambios en el ADN, como roturas o trasposiciones cromosómicas en las que los cabos rotos de dos cromosomas pueden intercambiarse. La radiación actúa como un iniciador de la carcinogénesis, induciendo alteraciones que progresan hasta convertirse en cáncer después de un período de latencia de varios años. Los rayos ultravioletas del sol y los rayos X aumentan la propensión a adquirir cáncer de piel y leucemia.

27
Infecciones o virus Existen cada vez más evidencias de que algunas infecciones pueden llegar a provocar cáncer. Se ha relacionado la bacteria Helicobacter pylori con el cáncer de estómago. 4 veces mas de pocibilidad de presentar cancer. El virus de Epstein-Barr se asocia con el linfoma de Burkitt y los linfoepiteliomas. El virus de la hepatitis con el hepatocarcinoma. El virus herpes tipo II o virus del herpes genital con el carcinoma de cérvix. Todos estos virus asociados a tumores humanos son del tipo ADN.

28
Estos virus del tipo ARN contienen un gen denominado oncogén viral, capaz de transformar las células normales en células malignas Los oncogenes virales tienen una contrapartida en las células humanas normales: es el protooncogén, u oncogén celular. Los productos de los oncogenes son factores de crecimiento que estimulan el crecimiento de las células tumorales.

29
Cáncer hereditario características propias
En el campo de la Genética Molecular, hoy se han comprobado una serie no despreciable de genes directamente involucrados en el desarrollo de diferentes tumores malignos hereditarios. La predisposición genética al cáncer implica que un gran número de personas tengan un riesgo mayor a consecuencia de su historia familiar y/o personal. El 5% de todas las neoplasias humanas son de carácter hereditario y en su mayoría siguen un modelo de herencia aparentemente dominante. Aparecen en edades más precoces que lo habitual para ese tipo de tumor. Historia familiar de cáncer del mismo tipo histológico en parientes de primer y segundo grados. Tumor multicéntrico en órganos únicos, y bilateral uni o multricéntrico en órganos pares. Aparición de más de un tumor primario en la misma persona. Aparición de tumores que coinciden con rasgos dismórficos o anomalías congénitas

30
Carcinogénesis El cáncer comienza en una célula, es decir que es de origen monoclonal. Esa célula alterada escapa a los controles y se vuelve “anarquica” iniciando una generación de más “células anárquicas”, que, a su vez, pueden inducir a cambios similares en las células vecinas.

31
Etapas de la carcinogénesis y acción de los carcinógenos
1. La INICIACIÓN: ocurre a nivel del genoma y las alteraciones pueden darse en los tumores benignos y malignos. Los agentes que actúan en la primer etapa pueden ser: fisicos, químicos o virales. 2.La PROMOCIÓN: la etapa de crecimiento tisular con la formación del tumor. Participan: los factores de crecimiento y los receptores a los factores de crecimiento, como así también la angiogénesis y degradación de las matrices extracelulares. 3. La PROGRESIÓN: implica la capacidad de invadir tejidos vecinos o a distancia por parte de la célula tumoral maligna. Esa capacidad está codificada también en los genes de la misma con modificaciones estructurales y funcionales.

32
Sólo una célula de entre diez mil que logre introducirse al torrente sanguíneo o linfático podrá asentarse para desarrollar un foco metastático. La célula maligna debe desprenderse de sus vecinas y “navegar” por el espacio intercelular y atravesar la membrana basal (degradación de matrices). Debe introducirse al vaso sanguíneo o linfático (migración celular). Debe sobrevivir al ataque de la respuesta inmune. Debe atravesar nuevamente la pared vascular y “anidar” en otro tejido que muchas veces no comparte su estirpe (colonización metastásica).
. Debe introducirse al vaso sanguíneo o linfático (migración celular). Debe sobrevivir al ataque de la respuesta inmune. Debe atravesar nuevamente la pared vascular y anidar en otro tejido que muchas veces no comparte su estirpe (colonización metastásica).")
33
HEMOGLOBINURIA PAROXÍSTICA
Síndrome de Marchiafava-Micheli Es una enfermedad adquirida y clonal del stem cell hematopoyético con la consiguiente producción de células sanguíneas defectuosas. Es una enfermedad poco frecuente. Afecta a los dos sexos y aparece a cualquier edad, aunque es más frecuente en adultos del sexo femenino.

34
En la fisiopatología de la HPN coexisten dos factores:
El fallo de la médula ósea normal, con una mutación somática del gen PIG-A. Cuando ambos factores ocurren en el mismo individuo, el clon HPN puede proliferar y el cuadro clínico de la enfermedad se hace evidente. Este clon anormal puede tener alguna ventaja proliferativa sobre el clon de células normales y hacerse dominante en la médula de estos pacientes.

35
Teoría de la patogénesis dual para el desarrollo de la HPN.
Se ha planteado la hipótesis de que el fallo de la médula ósea favorece el desarrollo del clon HPN, el cual se expande como resultado de una selección negativa contra el stem cell hematopoyético normal. En consecuencia, la mayoría de la hematopoyesis consistirá en células deficientes en proteínas ligadas al GPI. Teoría de la ventaja relativa del crecimiento o teoría de escape La cual se basa en el concepto de que la expansión del clon HPN depende de la existencia de uno o más factores ambientales adicionales externos, los cuales ejercen una presión selectiva a favor del clon HPN. Uno de estos factores podría ser una injuria a las células hematopoyéticas normales, lo cual salva a las células HPN anormales. Se supone que la injuria a la hematopoyesis ocurre a través del mecanismo del GPI.

36
Defecto en la biosíntesis del glicosilfosfatidilinositol (GPI)
La biosíntesis del GPI es defectuosa, debido a una mutación somática en el gen PIG-A localizado al final del brazo corto del cromosoma X. Estas alteraciones en el gen PIG-A afectan sólo a las células somáticas, específicamente a las células hematopoyéticas, por lo que se trata de una alteración adquirida, no hereditaria.

38
Deficiencia de la expresión de las proteínas de membrana
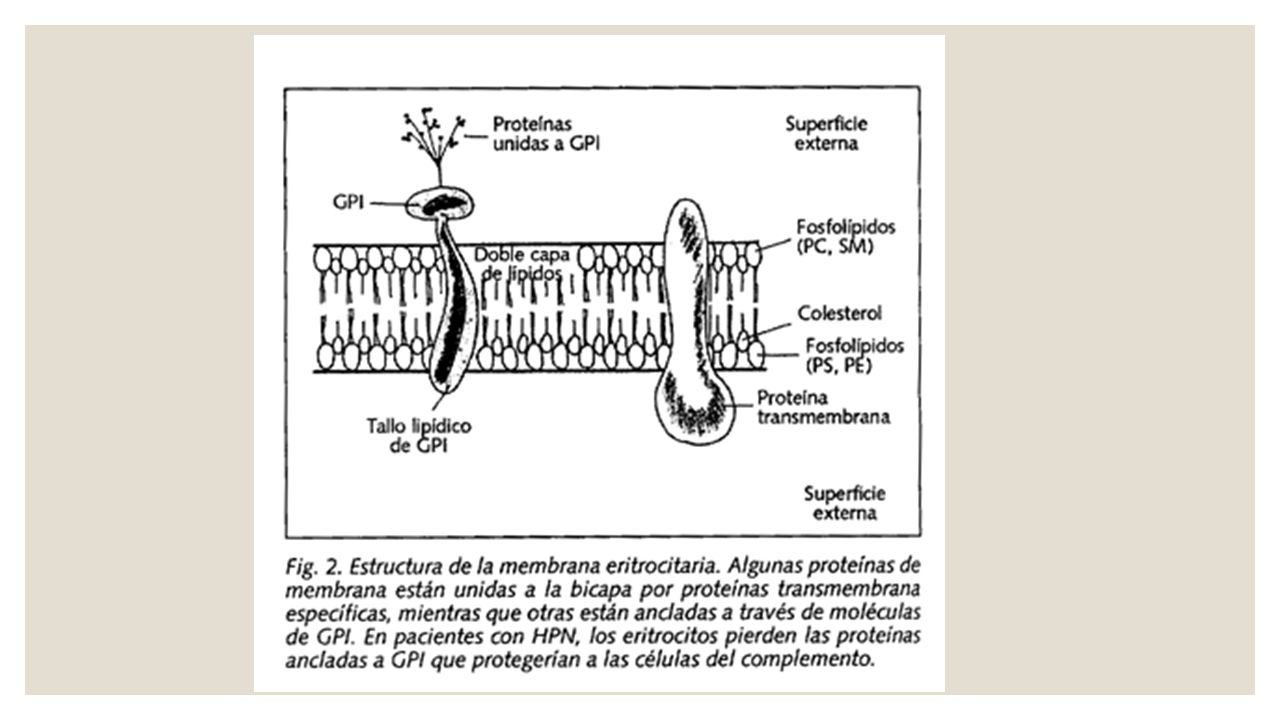
Disminución de la acetil colinesterasa en los hematíes y de la fosfatasa alcalina en los leucocitos. La ausencia de tres proteínas en la membrana del hematíe: CD55 o decay accelerating factor (DAF) que previene la formación de C3 convertasa. CD59 o inhibidor de la lisis reactiva de membrana [MIRL] El CD16 (receptor Fc g IIIa) es una proteína que se expresa en la superficie de los neutrófilos.
 que previene la formación de C3 convertasa. CD59 o inhibidor de la lisis reactiva de membrana [MIRL] El CD16 (receptor Fc g IIIa) es una proteína que se expresa en la superficie de los neutrófilos.")
39
Sensibilidad a la lisis mediada por complemento
La hemólisis de la HPN deriva de una alteración intrínseca de los glóbulos rojos. La destrucción de los eritrocitos es prematura, porque son muy susceptibles a la lisis mediada por complemento. HPN I: caracterizado por sensibilidad normal o casi normal al complemento. HPN II: Con ausencia parcial de CD 59, es de sensibilidad intermedia, de tres a cinco veces mayor que en las células normales. HPN III: Con ausencia completa de CD 59, es de 15 a 20 veces más susceptible a la lisis.

40
Manifestaciones clínicas
El patrón “clásico” de HPN se caracteriza por episodios de hemólisis intravascular que puede ser desde apenas detectable hasta masiva con requerimientos transfusionales y hemoglobinuria, que ocurren sobre todo asociados con el sueño y con una periocidad irregular. Al principio, el paciente refiere astenia, coloración amarillenta de la piel y otros síntomas de hemólisis crónica sin hemoglobinuria obvia Tratamiento En la HPN se han utilizado diferentes métodos terapéuticos, pero con excepción del trasplante de médula ósea, ninguno se considera apropiado. Desde el punto de vista práctico, el tratamiento de la HPN se divide en 3 aspectos fundamentales: a) Corrección de la anemia b) Prevención y tratamiento de la trombosis c) Modificación de la hematopoyesis.
 Corrección de la anemia. b) Prevención y tratamiento de la trombosis. c) Modificación de la hematopoyesis.")
41
PATOLOGÍAS DE CANALES PROTEICOS (CANALOPATIAS)
")
42
Las enfermedades asociadas a canales iónicos pueden ocurrir por diferentes razones. Por ejemplo:
a) por mutaciones cuyo resultado sea que se "pierde " la proteína o que es defectuosa. b) la proteína podría estar "sana", pero el sistema que la regula ser el anómalo. c) todo el sistema puede estar "sano", pero los canales son blanco de una amplia gama de toxinas que alteran su funcionalidad.
 por mutaciones cuyo resultado sea que se pierde la proteína o que es defectuosa. b) la proteína podría estar sana , pero el sistema que la regula ser el anómalo. c) todo el sistema puede estar sano , pero los canales son blanco de una amplia gama de toxinas que alteran su funcionalidad.")
43
La mayoría de las enfermedades que se han descrito como canalopatías son mutaciones genéticas. Por ejemplo, la fibrosis quística, enfermedad genética que afecta especialmente a los caucásicos, recesiva asintomática para los heterocigotos, pero de baja sobreviviencia en los homocigotos. Esta enfermedad se debe a defectos funcionales de los canales de cloruro presentes en intestino, páncreas, glándulas salivales, etc. Otras enfermedades

44
Fibrosis quística. Es una enfermedad que provoca la acumulación de moco espeso y pegajoso en los pulmones, el tubo digestivo y otras áreas del cuerpo. Es uno de los tipos de enfermedad pulmonar crónica más común en niños y adultos jóvenes. Es una enfermedad potencialmente mortal. Causas: causada por un gen defectuoso que lleva al cuerpo a producir un líquido anormalmente espeso y pegajoso llamado moco. Este moco se acumula en las vías respiratorias de los pulmones y en el páncreas. Síntomas: Los síntomas en los recién nacidos pueden abarcar: Retraso en el crecimiento. Incapacidad para aumentar de peso normalmente durante la niñez. Ausencia de deposiciones durante las primeras 24 a 48 horas de vida. Piel con sabor salado.

45
Tos o aumento de la mucosidad en los senos paranasales o los pulmones.
Fatiga. Congestión nasal causada por los pólipos nasales. Episodios recurrentes de neumonía (los síntomas de neumonía en una persona con fibrosis quística abarcan fiebre, aumento de la tos y dificultad respiratoria, aumento de la mucosidad y pérdida del apetito). Dolor o presión sinusal causados por infección o pólipos. Tratamiento: Antibióticos para prevenir y tratar infecciones sinusales y pulmonares. Vacuna antigripal y vacuna antineumocócica de polisacáridos (PPV, por sus siglas en inglés)
. Dolor o presión sinusal causados por infección o pólipos. Tratamiento: Antibióticos para prevenir y tratar infecciones sinusales y pulmonares. Vacuna antigripal y vacuna antineumocócica de polisacáridos (PPV, por sus siglas en inglés)")
46
Síndrome congénito Trastorno que se transmite de padres a hijos, en el cual un bebé presenta proteína en la orina e hinchazón del cuerpo. Congénito significa que está presente al nacer. Causas: Los niños que sufren este trastorno tienen una forma anormal de una proteína llamada nefrina. Los filtros de los riñones (glomérulos) necesitan esta proteína para funcionar de forma normal. Síntomas: Tos Disminución del gasto urinario Apariencia espumosa de la orina Bajo peso al nacer Inapetencia Hinchazón generalizada
 necesitan esta proteína para funcionar de forma normal. Síntomas: Tos. Disminución del gasto urinario. Apariencia espumosa de la orina. Bajo peso al nacer. Inapetencia. Hinchazón generalizada.")
47
Tratamiento: Antibióticos para controlar las infecciones.
Medicamentos para la presión arterial llamados inhibidores ECA y BRA para reducir la cantidad de proteína que se filtra en la orina. Diuréticos para eliminar el exceso de líquido. Antinflamatorios no esteroides (AINES), como indometacina, para reducir la cantidad de proteína que se filtra en la orina.
, como indometacina, para reducir la cantidad de proteína que se filtra en la orina.")
48
PATOLOGÍAS DE ENZIMAS

49
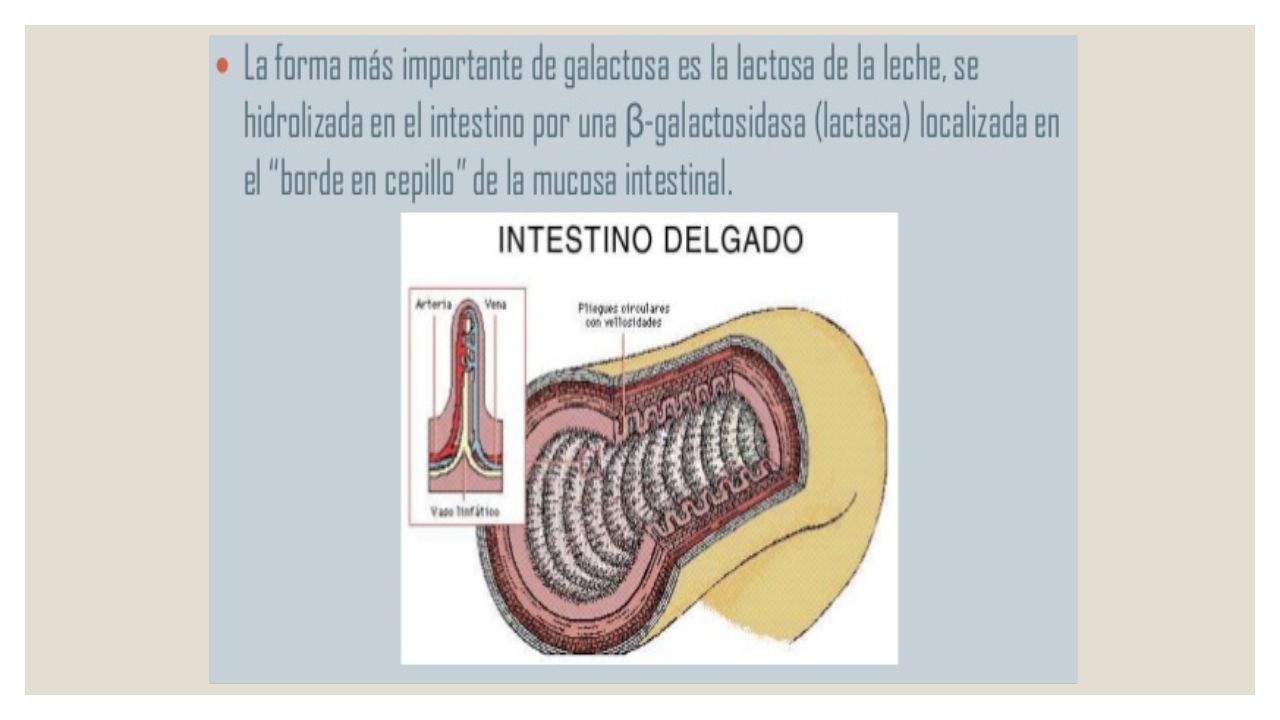
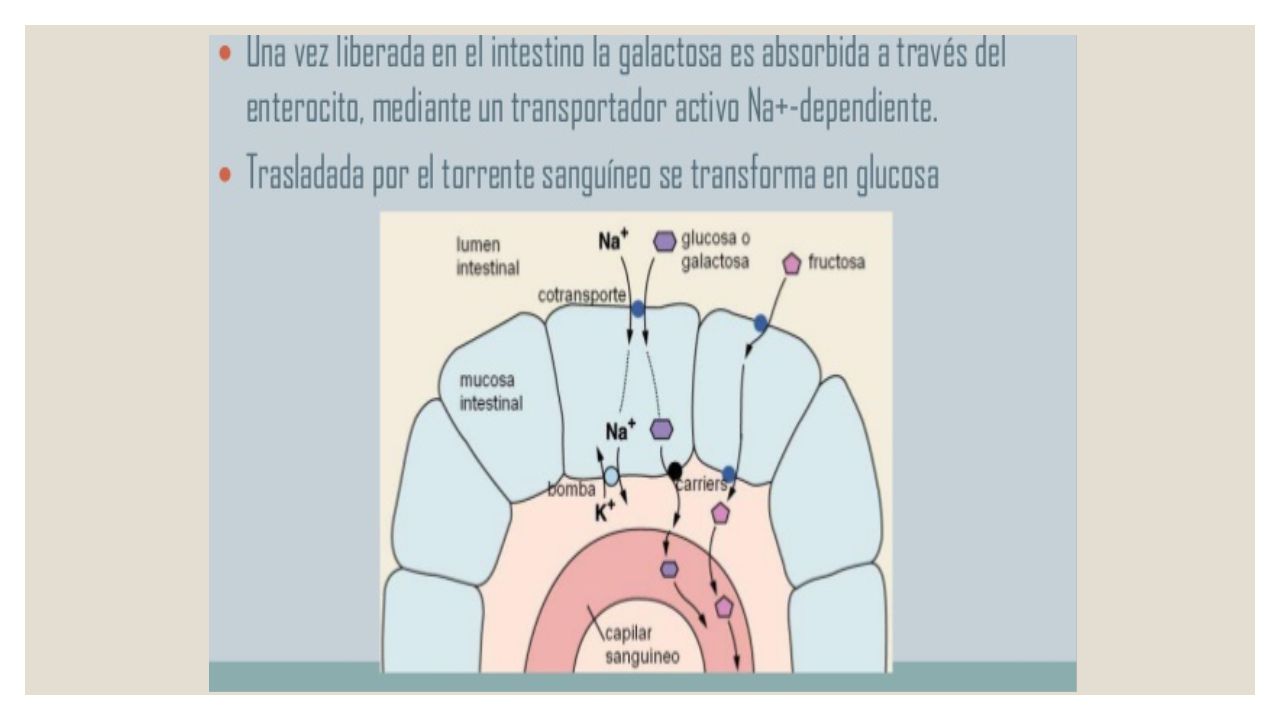
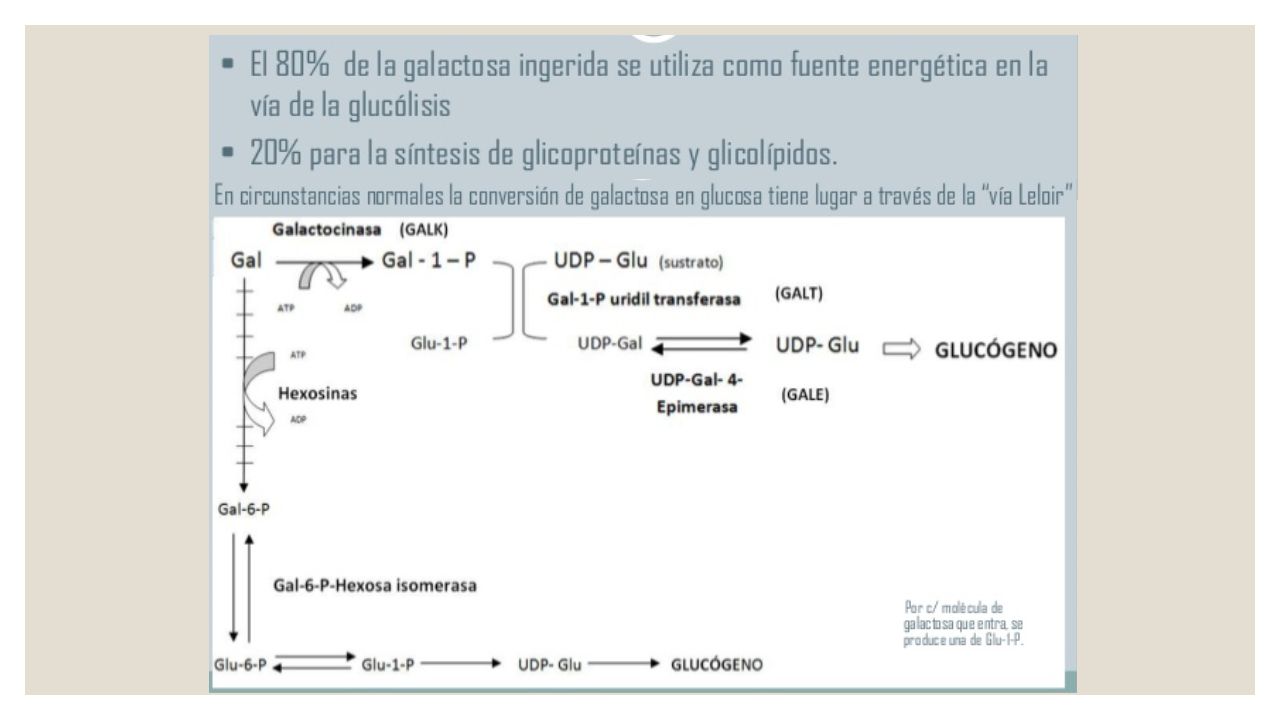
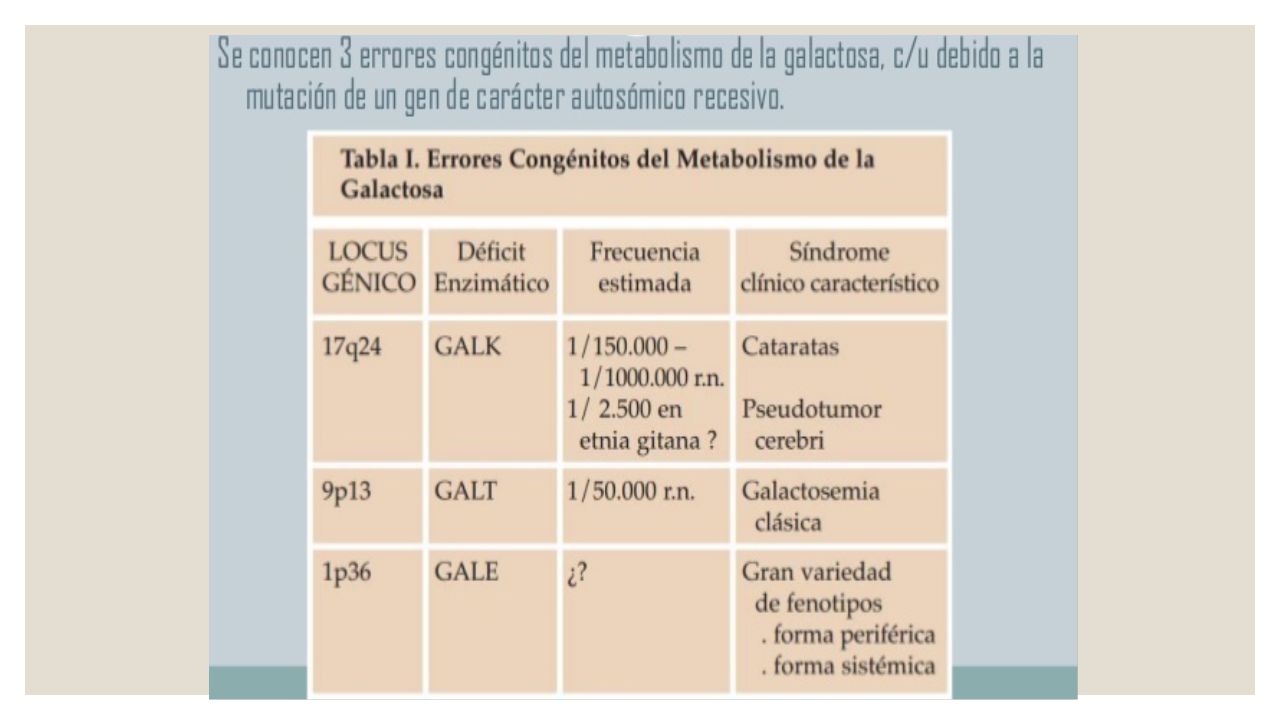
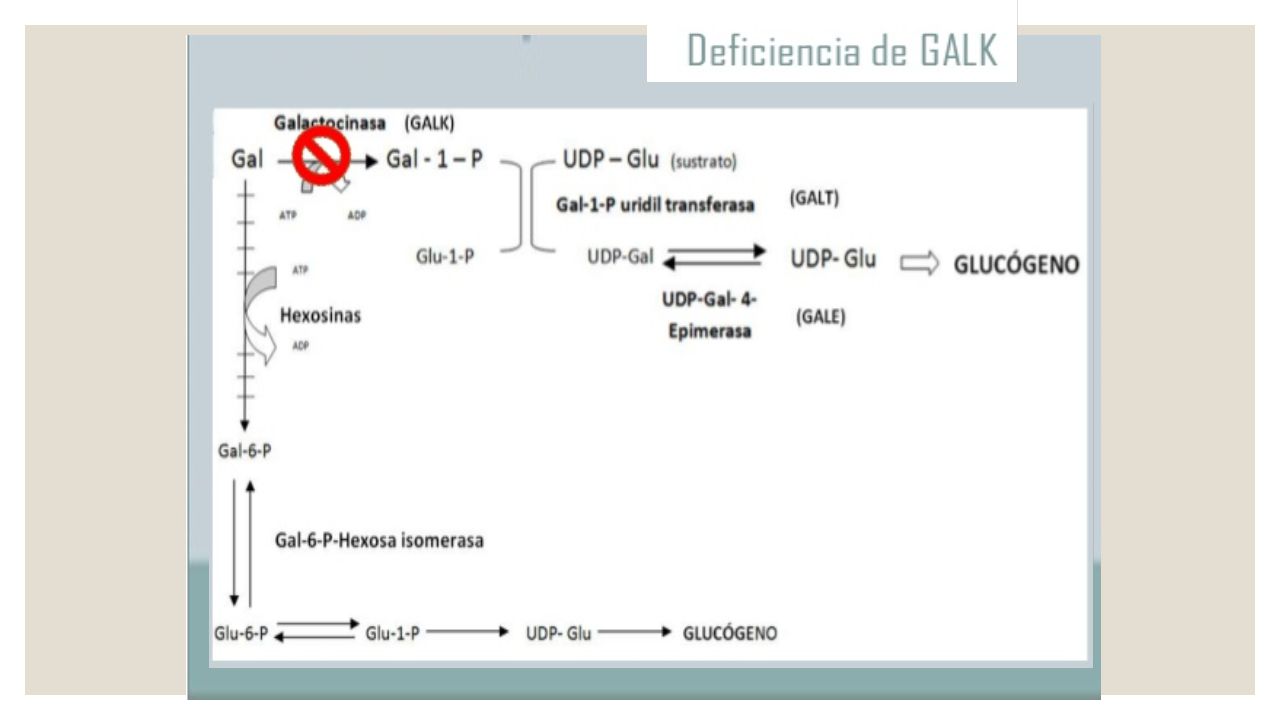
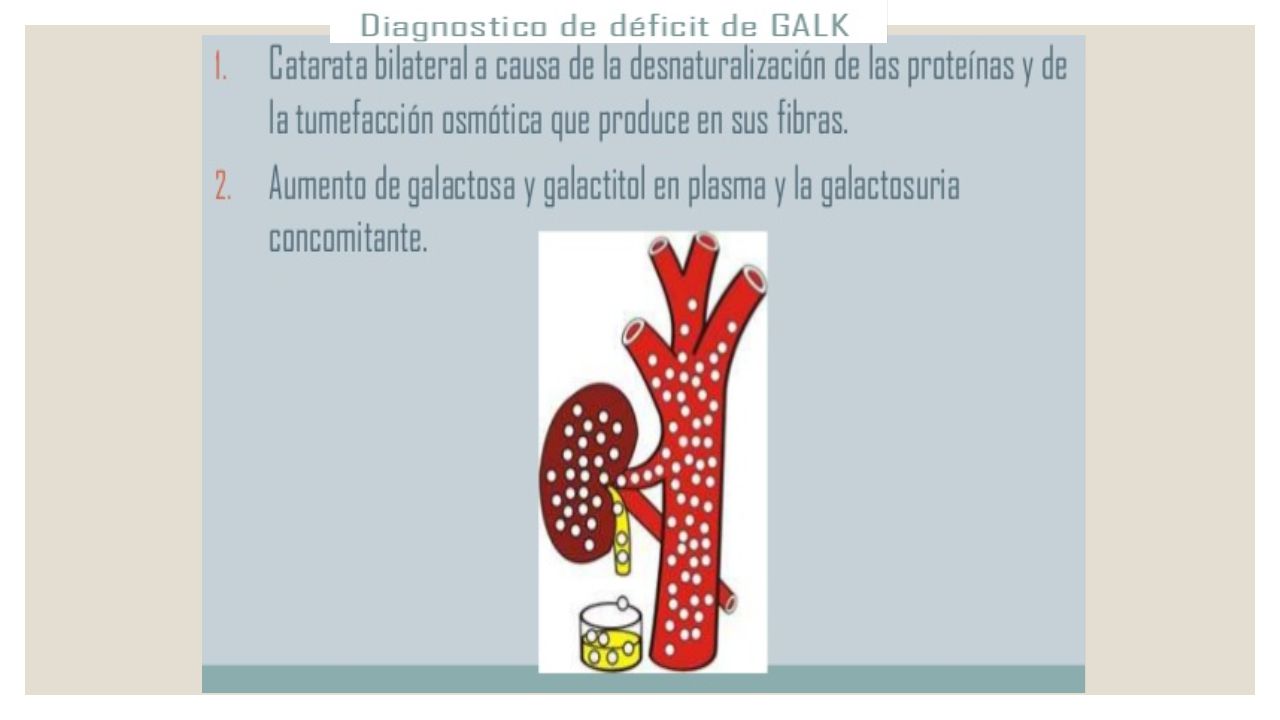
Galactosemia

74
Enfermedad de Wilson

75
Es un trastorno hereditario y presenta un patrón de herencia autosómico recesivo.

76
La sintomatología es muy diversa, por lo que el diagnóstico clínico de la enfermedad es complejo. Se caracteriza daños en el hígado, que puede ser desde la alteración de los niveles séricos de transaminasas hasta la cirrosis descompensada, incluyendo la hepatitis fulminante.

77
Los pacientes con esta enfermedad pueden presentar daños neurológicos, tales como: pérdida de memoria, dificultad de movimientos, temblores, párkinson, entre otros

78
Dentro de las enfermedades genéticas raras es tratable, sin embargo, de no atenderse de forma adecuada, puede provocar lesiones irreversibles en el hígado y el cerebro que pueden llevar a la muerte.

79
La causa molecular que la provoca son las mutaciones en el gen ATP7B (MIM ), el cual presenta 21 exones.
, el cual presenta 21 exones.")
80
Más de la mitad de las mutaciones son con pérdida de sentido dentro de los dominios transmembranales de la proteína ATP7B y en el lazo largo unidor de ATP, el resto está constituido por inserciones y deleciones pequeñas.2-10

Presentaciones similares