Descargar la presentación
La descarga está en progreso. Por favor, espere

1
Dra. Liliana P. Castro Zorrilla
Inmunodeficiencias Primarias (IDP) Dra. Liliana P. Castro Zorrilla Jefa Departamento Inmunología Instituto de Tisioneumonología Prof. Dr. Raúl Vaccarezza UBA- Hospital F. J. Muñiz 1
 Dra. Liliana P. Castro Zorrilla. Jefa Departamento Inmunología Instituto de Tisioneumonología Prof. Dr. Raúl Vaccarezza UBA- Hospital F. J. Muñiz. 1.")
2
INMUNODEFICIENCIAS PRIMARIAS SECUNDARIAS
Grupo heterogéneo de enfermedades que comprometen el desarrollo y las funciones del sistema inmune PRIMARIAS SECUNDARIAS

3
INMUNODEFICIENCIAS Secundarias o Adquiridas
*HIV SIDA *NO HIV Asociada a patología sistémica Metabólicas (Diabetes) Infecciosas Enfermedades neoplásicas Secundarias acción terapéutica Inmunosupresores Corticoides AINES Otras Drogas (anticonvulsivantes) Combinadas Trauma Cirugía Estrés psíquico y depresión (+ eje hipotálamo-hipofisario-adrenal y citoquinas proinflamatorias: IL-1, IL-6, TNF-, con desviación H2)
 Infecciosas. Enfermedades neoplásicas. Secundarias acción terapéutica. Inmunosupresores. Corticoides. AINES. Otras Drogas (anticonvulsivantes) Combinadas. Trauma Cirugía. Estrés psíquico y depresión. (+ eje hipotálamo-hipofisario-adrenal y. citoquinas proinflamatorias: IL-1, IL-6, TNF-, con desviación H2)")
4
INMUNODEFICIENCIAS Secundarias NO HIV
Asociadas a patología sistémica: Metabólicas: Malnutrición calórico proteica Deficiencia vitamínica/oligoelementos Diabetes mellitus Deficiencias enzimáticas Enfermedades renales Enteropatía perdedora de proteínas

5
INMUNODEFICIENCIAS Secundarias NO HIV
Asociadas a patología sistémica: Infecciosas Virus: sarampión, EBV, CMV, Herpes, HBV, HAV, HCV, influenza, rabia, rubéola, etc. Bacterias: Mycobacterium tuberculosis, entero/endotoxinas (SuperAg) Hongos: cándida, histoplasma Parásitos: histo/hemoparásitos (Tripanosoma cruzi) Enfermedades neoplásicas Linfoproliferativas: Hodgkin, leucemias
 Hongos: cándida, histoplasma. Parásitos: histo/hemoparásitos (Tripanosoma cruzi) Enfermedades neoplásicas. Linfoproliferativas: Hodgkin, leucemias.")
6
INMUNODEFICIENCIAS Secundarias NO HIV
Secundarias acción terapéutica Inmunosupresores Ciclosporina, tacrolimus, metrotexato, ciclofosfamida Corticoides AINES IgG Otras Drogas: Anticonvulsivantes, talidomida, aminoglucósidos, tetraciclinas

7
INMUNODEFICIENCIAS Secundarias NO HIV
Combinadas Trauma Cirugía Estrés físico agudo (descarga catecolaminas y cortisol) Ruptura barrera mecánica de piel y mucosas Alteraciones metabólicas/hidroelectrolíticas secundarias a injuria tisular Estrés psíquico y depresión
 Ruptura barrera mecánica de piel y mucosas. Alteraciones metabólicas/hidroelectrolíticas. secundarias a injuria tisular. Estrés psíquico y depresión.")
8
ESTRÉS Y SISTEMA INMUNE
Dura minutos u horas Es INMUNOESTIMULANTE Redistribución de leucocitos al “lugar correcto” (herida, infección) Estrés agudo Hipersensibilidad retardada (resp. celular) Respuesta humoral primaria y secundaria Respuesta inmune innata y adaptativa Dura muchas horas, días, semanas, o meses Es INMUNOSUPRESOR Estrés crónico Hipersensibilidad retardada (resp. celular) Producción de anticuerpos Actividad de los macrófagos Actividad de las células NK 8
 Estrés. agudo. Hipersensibilidad retardada (resp. celular) Respuesta humoral primaria y secundaria. Respuesta inmune innata y adaptativa. Dura muchas horas, días, semanas, o meses. Es INMUNOSUPRESOR. Estrés. crónico. Hipersensibilidad retardada (resp. celular) Producción de anticuerpos. Actividad de los macrófagos. Actividad de las células NK. 8.")
9
1998 Marshall Describió que ante el estrés crónico secundario a exámenes finales en estudiantes sanos→ desregulación del perfil de citoquinas hacia las H2 2002 Kilpelainen → estudiantes demostró como el estrés favorecía las manifestaciones de asma y conjuntivitis alérgicas. 2006 Kimata: niños expuestos a situaciones estresantes (videojuegos de combate 2hs o llamadas repetidas a un celular en 30’) ↑ el nivel de TH2, neuropéptidos y neurotrofinas 9
 ↑ el nivel de TH2, neuropéptidos y neurotrofinas. 9.")
10
SIDA Y ESTRES La rápida progresión de enfermedad: está relacionada al patrón de citoquinas H2: (secreción local noradrenalina que inhibe IL-12): caquexia, síndrome de desgaste metabólico, génesis de tumores de células B, infecciones EBV, demencia, etc Los que mejoran espontáneamente: buena respuesta CD8+, perfil citoquinas H1 10
: caquexia, síndrome de desgaste metabólico, génesis de tumores de células B, infecciones EBV, demencia, etc. Los que mejoran espontáneamente: buena respuesta CD8+, perfil citoquinas H")
11
DEPRESIÓN Perfil inmunológico, endócrino y bioquímico semejante al del estrés. Cursa con activación eje-hipotálamo-hipofisario-adrenal, y activación citoquinas proinflamatorias (IL-1, IL-6, TNF-) con desviación H2 11
 con desviación H")
12
INMUNODEFICIENCIAS PRIMARIAS (IDP)
Enfermedades que resultan de defectos primarios del sistema inmune Más de 180 entidades bien definidas La gran mayoría son hereditarias monogénicas Se han identificado más de 110 genes alterados Baja frecuencia: 1 en nacidos vivos

13
Tres Niveles de Defensa
BARRERRAS ANATÓMICAS Y FISIOLÓGICAS INMUNIDAD INNATA Lisozima en secreciones Complemento Integridad de la piel Neutrófilo INMUNIDAD ADQUIRIDA DC Ph bajo estómago Células T Células NK El sistema inmune consiste en una red de cooperación entre numerosas células y componentes solubles que le permite ejercer sus funciones de defensa frente a los microorganismos invasores. Los componentes se pueden clasificar por su origen en los de inmunidad innata, filogenéticamente anterior, y los de inmunidad adquirida, más reciente. Cuando las barreras anatómicas y fisiológicas son lesionadas o alteradas, se activan las respuestas de inmunidad innata y adquirida. Pertenecen a la inmunidad innata las células fagocíticas, las células asesinas naturales (del inglés, NK), el complemento, las citocinas y los péptidos antimicrobianos. La inmunidad adquirida produce respuestas altamente específicas dirigidas al germen invasor mediante la activación de los linfocitos T y B, producción de anticuerpos, y secreción de citocinas y quimiocinas. El nivel óptimo de inmunidad se alcanza con la cooperación entre la inmunidad innata y la adquirida, y cualquier falla en sus componentes puede tener consecuencias clínicas. Presencia de comensales Células B Clearance ciliar y mucus de las vías respiratorias 13
, el complemento, las citocinas y los péptidos antimicrobianos. La inmunidad adquirida produce respuestas altamente específicas dirigidas al germen invasor mediante la activación de los linfocitos T y B, producción de anticuerpos, y secreción de citocinas y quimiocinas. El nivel óptimo de inmunidad se alcanza con la cooperación entre la inmunidad innata y la adquirida, y cualquier falla en sus componentes puede tener consecuencias clínicas. Presencia de comensales. Células B. Clearance ciliar y mucus de las vías respiratorias. 13.")
14
Inmunodeficiencias Primarias
Tipo de célula afectada Etapa del desarrollo en la que ocurre el defecto

15
Inmunodeficiencias Primarias
Defectos en la activación T y/o B

16
Inmunodeficiencias Primarias

17
Inmunodeficiencias Primarias

18
EPIDEMIOLOGÍA: IDP Etapa temprana de la vida
40% durante 1° año de vida 95% antes de los 16 años. 60-80 % Hombres: ligadas cromosoma X Antecedentes familiares: infecciones severas, muertes tempranas (25% portadores fliares) Infecciones recidivantes, prolongadas, graves, complicadas, microorganismos no patógenos, en diferentes localizaciones y sistemas Tipo de gérmen: correlación con tipo de ID
 Infecciones recidivantes, prolongadas, graves, complicadas, microorganismos no patógenos, en diferentes localizaciones y sistemas. Tipo de gérmen: correlación con tipo de ID.")
19
IDP EDAD DE PRESENTACIÓN 0 - 1 año 40 % 1 - 5 años 40 %

20
Inmunodeficiencias Primarias
Mundial: 10 millones de casos. Argentina:: 2005: 1319 pacientes (Registro Nacional de la SAP) Humorales: 69,5 % (918 ptes) Celulares: 3,94% (52 (Ptes) Déficit selectivo de IgA : 561 ptes Inmunodeficiencia Común Variable: IDCV: 127 ptes Instituto Vaccarezza ( ) 19 pacientes Inmunodeficiencia Común Variable (11 sexo masculino y 8 sexo femenino, 7 fallecidos) 21 pacientes Déficit Selectivo IgA
 Humorales: 69,5 % (918 ptes) Celulares: 3,94% (52 (Ptes) Déficit selectivo de IgA : 561 ptes. Inmunodeficiencia Común Variable: IDCV: 127 ptes. Instituto Vaccarezza ( ) 19 pacientes Inmunodeficiencia Común Variable. (11 sexo masculino y 8 sexo femenino, 7 fallecidos) 21 pacientes Déficit Selectivo IgA.")
21
Argentina Registro Latinoamericano de IDP

22
REGISTRO LASID OCTUBRE 2011 Argentina 38 % de casos Agamma XL
(2005: 1319 ptes 69,5% Def humorales) IDCV 17,26 % Déf selectivo IgA 17,45%
 IDCV 17,26 % Déf selectivo IgA. 17,45%")
23
CLASIFICACIÓN DE IDP Las principales categorías son:
I. Deficiencias predominantemente de anticuerpos (>50%) II. Inmunodeficiencias Combinadas T y B (10-20%) III. Otros Síndromes de Inmunodeficiencia bien definidos (5-10%) IV. Defectos congénitos del número y/o función del fagocito (15-20%) V. Deficiencias del Sistema Complemento (2%) VI. Enfermedades por desregulación inmune VII. Defectos en la inmunidad innata VIII. Desórdenes autoinflamatorios
 II. Inmunodeficiencias Combinadas T y B (10-20%) III. Otros Síndromes de Inmunodeficiencia bien definidos (5-10%) IV. Defectos congénitos del número y/o función del fagocito (15-20%) V. Deficiencias del Sistema Complemento (2%) VI. Enfermedades por desregulación inmune. VII. Defectos en la inmunidad innata. VIII. Desórdenes autoinflamatorios.")
24
IDP Mayor susceptibilidad a las infecciones Antecedentes familiares
Manifestaciones Mayor susceptibilidad a las infecciones Mayor incidencia de malignidad Mayor incidencia de autoinmunidad/inflamación Manifestaciones NO inmunológicas Reacciones adversas a vacunas (SABIN BCG) RN: infección y tiempo de caída cordón umbilical Antecedentes familiares Muertes neonatales no explicadas. Familiares con ID Consanguinidad Factores de riesgo VIH Patrón herencia: AD, AR, LX
 RN: infección y tiempo de caída cordón umbilical. Antecedentes familiares. Muertes neonatales no explicadas. Familiares con ID. Consanguinidad. Factores de riesgo VIH. Patrón herencia: AD, AR, LX.")
25
IDP Inflamación Aftas orales Eccemas Dermatitis seborreica
Artritis inflamatoria Enfermedad intestinal Sarcoidosis like Meningitis Maliginidad 4-40% Linfomas Leucemias Carcinomas Autoinmunidad Citopenias PTI AHA Coombs+ Neutropenia autoinmune Enfermedad celíaca Endocrinopatías, Hipotiroidismo Vasculitis No inmunológicas Cardíacas. cabello azúl Deformaciones óseas, neurológicas

26
IDP Examen físico Detención crecimiento: estatura, peso de acuerdo a percentilos. Presencia o no de tej linfoideo: amígdalas, ganglios periféricos. Hepatoesplenomegalia Abscesos recurrentes Telangiectasias Marcha atáxica Albinismo

27
IDP: Interrogatorio Manifestaciones Infecciosas: Localización
Frecuencia (recurrencia, cronicidad) Mayor gravedad, duración Evolución, respuesta tratamiento Complicaciones, secuelas Microorganismos
 Mayor gravedad, duración. Evolución, respuesta tratamiento. Complicaciones, secuelas. Microorganismos.")
28
IDP: INFECCIONES RECURRENTES VÍAS RESPIRATORIAS
DIAGNÓSTICOS DIFERENCIALES 1-Malformaciones Congénitas Quiste Broncogénico- Secuestro Pulmonar 2-Déficit -1 antitripsina 3-Fibrosis Quística (50% de Bronquiectasias) 4-Disquinesia Ciliar 5-Inmunodeficiencia Primaria
 4-Disquinesia Ciliar. 5-Inmunodeficiencia Primaria.")
29
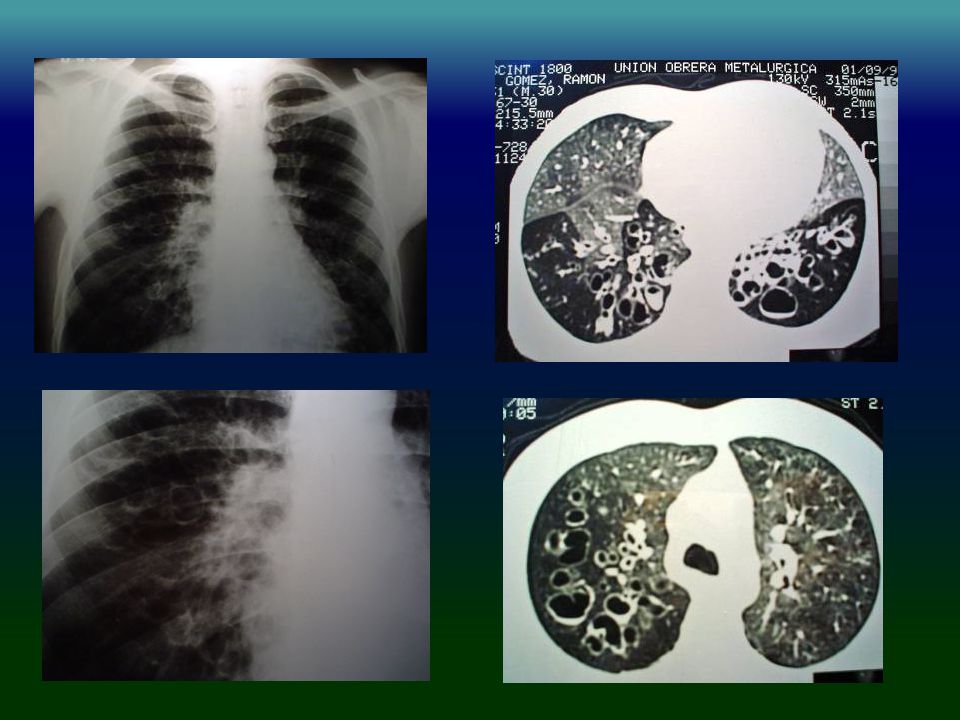
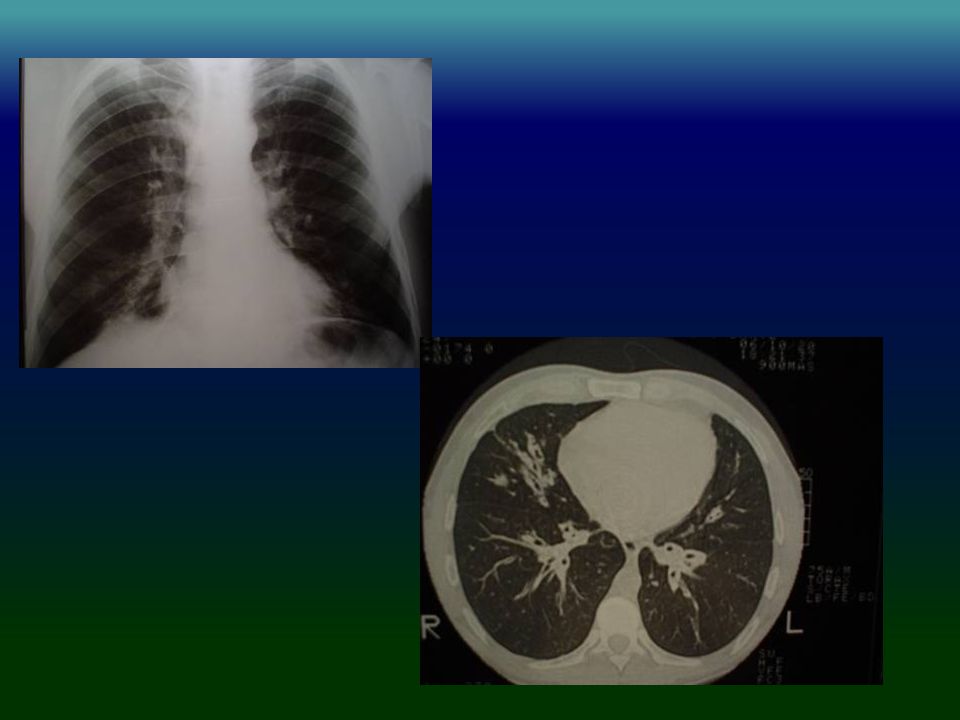
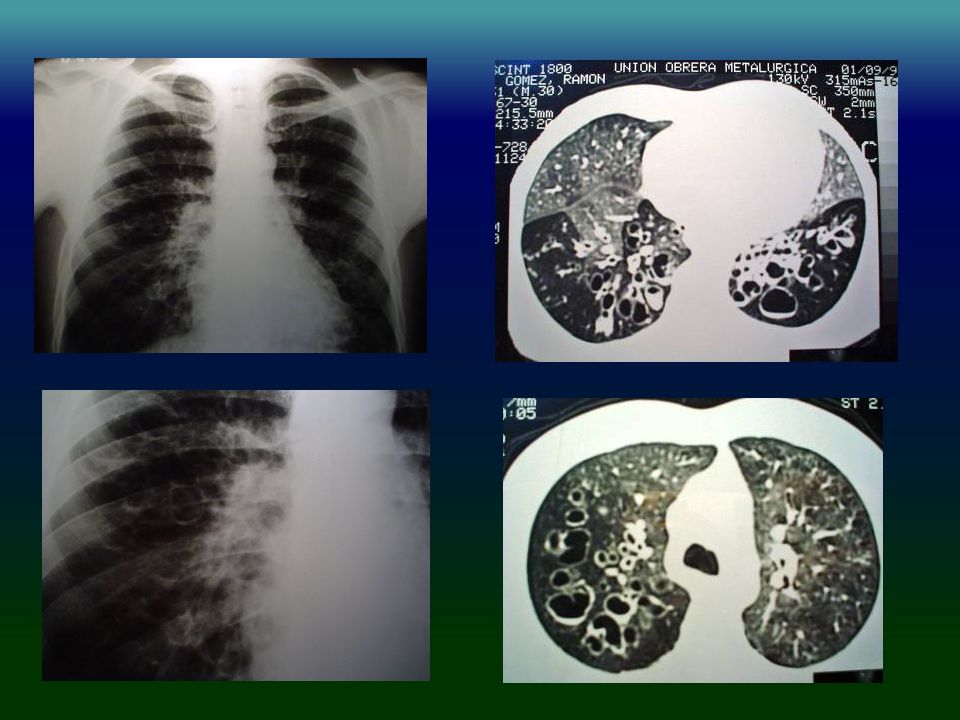
IDP: PATRÓN RADIOLÓGICO
Engrosamiento pared bronquial % Bronquiectasias % Engrosamiento pleural % Atelectasias % Cavidades % Bullas % Obregon RG Chest 1994,106:

30
IDCV Dra Liliana Castro Zorrilla

31
IDP: TAC HRCT Engrosamiento pared bronquial 90%
Bronquiectasias cilíndricas % Opacidades en vidrio esmerilado % Atrapamiento aéreo % Obregon RG Chest 1994,106:

33
HISTORIA CLÍNICA : IDP Aparato digestivo Diarrea crónica (Giardias)
Sindrome de malabsorción Retraso pondo-estatural

34
¿Cuando sospechar una Inmunodeficiencia Primaria ?

35
¿Cuántas infecciones deben ser consideradas
para pensar en una Inmunodeficiencia? > de 4-5 infecciones respiratorias/año >más de 1 infección por un patógeno inusual 1 infección prolongada puede dar indicio de mal funcionamiento del sistema inmune

36
Ocho o más infecciones óticas en menos de un año
Dos o más sinusitis graves en menos de un año. Dos meses a más de tratamiento con antibióticos con poco efecto

37
Dos o más neumonías en menos de un año.
Curvas de peso y talla inadecuadas en los niños. Abscesos en órganos internos y/o cutáneos profundos recurrentes Aftas bucales o lesiones cutáneas persistentes, después del primer año de vida.

38
Necesidad de administrar antibióticos EV
para erradicar las infecciones. Dos o más infecciones severas en un año. Historia familiar de alguna inmunodeficiencia primaria.

39
IDP: déficit predominante AC
Agammaglobulinemia LX (Enf Bruton) Sind Hiper IgM no ligado al sexo Deleción genes cadenas pesadas Ig Mutación cadena Kappa Déficit selectivo subclases de IgG Déficit de Ac con Igs normales Inmunodeficiencia Común Variable Deficit selectivo de IgA Hipogamma transitoria de la infancia Agammaglobulinemia autosómica recesiva
 Sind Hiper IgM no ligado al sexo. Deleción genes cadenas pesadas Ig. Mutación cadena Kappa. Déficit selectivo subclases de IgG. Déficit de Ac con Igs normales. Inmunodeficiencia Común Variable. Deficit selectivo de IgA. Hipogamma transitoria de la infancia. Agammaglobulinemia autosómica recesiva.")
40
Déficit selectivo de IgA:
1:700 en caucásicos 1: japoneses Patrón hereditario variable (asociado a haplotipos de HLA) Pacientes asintomáticos o con infecciones sinopulmonares, gastrointestinales, urinarias. IgA ausente o muy disminuída; IgG e IgM Normal Se supone defecto en la diferenciación de células B. Células T Normales. Asociación con patología autoinmune (LES, AR) enfermedad celíaca, asma y alergias Asociado a déficit selectivo subclases IgG2 e IgG4.
 Pacientes asintomáticos o con infecciones sinopulmonares, gastrointestinales, urinarias. IgA ausente o muy disminuída; IgG e IgM Normal. Se supone defecto en la diferenciación de células B. Células T Normales. Asociación con patología autoinmune (LES, AR) enfermedad celíaca, asma y alergias. Asociado a déficit selectivo subclases IgG2 e IgG4.")
41
Inmunodeficiencia Común Variable
Todas aquellas inmunodeficiencias humorales de presentación clínica tardía de la vida. La OMS la define como una entidad con “función deficiente de anticuerpo (condición sine qua non para el diagnóstico), generalmente acompañada de hipogammaglobulinemia y ocasional compromiso celular T” “TARDIA” desarrollo posterior 24 meses de vida; lo habitual es presentación en adultos jóvenes “COMÚN” por su frecuencia “VARIABLE” en cuanto a grado de inmunodeficiencia humoral y cuadros clínicos 1 cada nacidos vivos
, generalmente acompañada de hipogammaglobulinemia y ocasional compromiso celular T TARDIA desarrollo posterior 24 meses de vida; lo habitual es presentación en adultos jóvenes. COMÚN por su frecuencia. VARIABLE en cuanto a grado de inmunodeficiencia humoral y cuadros clínicos. 1 cada nacidos vivos.")
42
IDCV: etiopatogenia Componente genético, por lo que se considera una IDP, aunque no se conoce su patrón preciso de herencia. Las células B no maduran hasta convertirse en células plasmáticas. Bloqueo de la maduración de células B hasta la etapa de células plasmáticas o incapacidad de éstas para producir las formas secretorias de inmunoglobulinas. Mutaciones: ICOS, TACI, BAFF-R, CD19.

43
IDCV: Infecciones no asociada al sexo y presentación clínica tardía
en la susceptibilidad a las infecciones. sinusopulmonares (sinusitis, otitis, bronquitis y neumonías) por Streptococcus pneumoniae, Haemophilus influenzae, Klebsiella pneumoniae., infección por micobacterias, hongos y Pneumocystis jiroveci. herpes simple e infecciones graves por citomegalovirus septicemia e infecciones recurrentes en la piel, articulaciones, aparato urinario y sistema nervioso meningoencefalitis de evolución tórpida y desenlace fatal asociado a infección por echovirus
 por Streptococcus pneumoniae, Haemophilus influenzae, Klebsiella pneumoniae., infección por micobacterias, hongos y Pneumocystis jiroveci. herpes simple e infecciones graves por citomegalovirus. septicemia e infecciones recurrentes en la piel, articulaciones, aparato urinario y sistema nervioso. meningoencefalitis de evolución tórpida y desenlace fatal asociado a infección por echovirus.")
44
IDCV: Manifestaciones gastrointestinales
20% infección e inflamación asociado a síndrome de malabsorción . 10% de infecciones por Salmonella, Campylobacter, o Giardia lambia . 40% de infección por H. pylori , asociada a dispepsia (ausencia IgA mucosa) 5% la inflamación GI simula una enfermedad de Crohn o una colitis ulcerosa (2%)
 5% la inflamación GI simula una enfermedad de Crohn o una colitis ulcerosa (2%)")
45
IDCV: Manifestaciones autoinmunes
20 a 25% desarrollan uno o más fenómenos autoinmunes: artritis reumatoidea, LES hasta una variedad de citopenias, hepatitis, vitiligo y alopecía areata 4-5 % tromobocitopenia idiopática y anemia hemolítica. 6% tienen varias enfermedades autoinmunes e incluso se han descrito síndromes poliautoinmunes . 8-20% granulomas no caseosos (sarcoid-like)
")
46
IDCV: neoplasias 8%:linfoma Hodgkin
24 diferentes tumores, inlcuyendo cáncer de mama, de próstata, carcinoma de células escamosas, melanoma y carcinoma de células basales. 30 a 400 veces mayor el riesgo a desarrollar hiperplasia nodular linfoide (HNL). 50 veces más: ca gástrico
. 50 veces más: ca gástrico.")
47
IDCV: diagnóstico Laboratorio:
nivel sérico de las concentraciones de IgG, rangos reducidos (< 100 mg/dL) hasta justo por debajo del rango normal de mg/dL
 hasta justo por debajo del rango normal de mg/dL.")
48
IDCV: diagnóstico Nivel I Hemograma, proteinograma.
Determinación sérica de IgG, IgM, IgA e IgE (estudio cuantitativo) Búsqueda de anticuerpos preexistentes: Isohemaglutininas (antiA y antiB), Anti-estreptolisona O (ASTO), Anti-toxina tetánica, Anti-toxoide diftérico. Recuento de Linfocitos B por expresión de antígenos de diferenciación tales como CD19, o CD20 (estudio cuantitativo)
 Búsqueda de anticuerpos preexistentes: Isohemaglutininas (antiA y antiB), Anti-estreptolisona O (ASTO), Anti-toxina tetánica, Anti-toxoide diftérico. Recuento de Linfocitos B por expresión de antígenos de diferenciación tales como CD19, o CD20 (estudio cuantitativo)")
50
IDCV: diagnóstico Nivel II
Respuesta de anticuerpos a la inmunización activa con toxoides tetánico, diftérico, polisacáridos de neumococo, hepatitis, sarampión (pruebas funcionales). Producción de inmunoglobulinas in vitro mediante estimulación con mitógenos tales como el PWM (prueba funcional). Determinación de subclases de IgG: IgG1, IgG2, IgG3, IgG4 (estudio cuantitativo).
. Producción de inmunoglobulinas in vitro mediante estimulación con mitógenos tales como el PWM (prueba funcional). Determinación de subclases de IgG: IgG1, IgG2, IgG3, IgG4 (estudio cuantitativo).")
51
DIAGNÓSTICO IDP Evaluación RI celular Linfocitos/mm3
Dosaje LT: CD4 CD8 CD3 RHR (PPD, candidina, tricofitina) Cultivos Dosajes citoquinas Actividad enzimática (ADA)
 Cultivos. Dosajes citoquinas. Actividad enzimática (ADA)")
52
Agamaglobulinemia Congénita de Bruton o ligada al X
1 cada nacimientos Identificada en 1952 Enfermedad ligada al cromosoma X, mutación puntual en el gen de la tirosina quinasa de Bruton (BTK) Bloqueo de la maduración de linfocitos pre-B a linfocitos B con Ig M de superficie Ausencia o de: centros germinales en ganglios linfáticos células plasmáticas en tejidos LB (<2%) sangre periférica y tejidos linf Inmunoglobulinas IgG < 200 mg% CARACTERISTICAS: artritis crónica, meningoencefalitis enterovirus, dermatomiositis.
 Bloqueo de la maduración de linfocitos pre-B a linfocitos B con Ig M de superficie. Ausencia o de: centros germinales en ganglios linfáticos. células plasmáticas en tejidos. LB (<2%) sangre periférica y tejidos linf. Inmunoglobulinas IgG < 200 mg% CARACTERISTICAS: artritis crónica, meningoencefalitis enterovirus, dermatomiositis.")
53
Agamaglobulinemia Congénita de Bruton o ligada al X
Ausencia total de inmunoglobulinas séricas. Ligada a cromosoma X, afecta sólo a varones. Infecciones recidivantes y graves desde los 8 o 9 meses de edad. Patógenos piógenos (estafilococos, Haemophilus influenzae, etc). Conjuntivitis, faringitis, otitis, bronquitis, neumonías, infecciones cutáneas). Otras enfermedades asociadas (virales, y autoinmunitarias). Ganglios linfáticos y bazo con ausencia centros germinales. Ausencia de células plasmáticas en ganglios, bazo, médula ósea y tejidos. Amígdalas mal desarrolladas. 53
. Conjuntivitis, faringitis, otitis, bronquitis, neumonías, infecciones cutáneas). Otras enfermedades asociadas (virales, y autoinmunitarias). Ganglios linfáticos y bazo con ausencia centros germinales. Ausencia de células plasmáticas en ganglios, bazo, médula ósea y tejidos. Amígdalas mal desarrolladas. 53.")
54
Gammaglobulina parenteral en el tratamiento de las IDP
54

55
Gammaglobulina Endovenosa (GGEV)
Producto derivado pool plasmático de miles de donantes. Contiene diversidad de moléculas de IgG elaboradas en respuesta a a) agentes infecciosos prevalentes de una región b) a vacunaciones. Logrando cubrir el mayor margen posible de acciones fisiológicas de las Inmunoglobulinas: efectos antiinfecciosos, regulación inmunológica, red idiotípica, etc De c/litro plasma se obtienen 2-3 g de Igs (para 25 g Inmunoglobulina se necesitan: litros plasma) a donantes 55
 agentes infecciosos prevalentes de una región. b) a vacunaciones. Logrando cubrir el mayor margen posible de acciones fisiológicas de las Inmunoglobulinas: efectos antiinfecciosos, regulación inmunológica, red idiotípica, etc. De c/litro plasma se obtienen 2-3 g de Igs. (para 25 g Inmunoglobulina se necesitan: litros plasma) a donantes. 55.")
56
GGEV: Mecanismo de acción
Actividad antígeno específica: posee Ac con 10 millones de especificidades diferentes cumplen funciones de opsonización, neutralización y activación del complemento Acción inmunomoduladora Solubilización de IC’ circulantes y depositados en tejidos. Bloquea receptor Fc Acción inmunomoduladora, a través de: Inhibición de la producción y liberación de citoquinas por macrófagos y linfocitos T: IL-1, IL-2, IL-3, IL-4, IL-5, IL-6, IL-10, GM-CSF, TNF alfa, IFN gamma Aumenta la producción de IL-8 y del antagonista del receptor de IL-1 (IL-1 Ra) Tiene anticuerpos contra citoquinas (IL-1, TNF alfa) Disminución de la expresión del receptor para IL-2 Inhibe la activación de los linfocitos T y B, y disminuye la producción de inmunoglobulinas por los linfocitos B Inhibe la activación de los linfocitos T mediada por superantígenos Ejerce interacciones reguladoras idiotipo/anti idiotipo en la red idiotípica Bloquea los receptores para el Fc gamma presentes en macrófagos, linfocitos T y B Ayuda a la solubilización de complejos inmunes circulantes y depositados en tejidos. 56
 Tiene anticuerpos contra citoquinas (IL-1, TNF alfa) Disminución de la expresión del receptor para IL-2. Inhibe la activación de los linfocitos T y B, y disminuye la producción de inmunoglobulinas por los linfocitos B. Inhibe la activación de los linfocitos T mediada por superantígenos. Ejerce interacciones reguladoras idiotipo/anti idiotipo en la red idiotípica. Bloquea los receptores para el Fc gamma presentes en macrófagos, linfocitos T y B. Ayuda a la solubilización de complejos inmunes circulantes y depositados en tejidos. 56.")
57
Uso de la gammaglobulina
Sustitutivo como terapia de reemplazo: Inmunodeficiencias Primarias Inmunodeficiencias Secundarias Anti-inflamatorio / inmunomodulador: Enfermedades autoinmunes Enfermedades inflamatorias

58
GGEV: Indicaciones IDP/ID Humoral terapia reemplazo: Agammag, IDCV, Sind Hiper IgM, Def AC antipolisacáridos, Sind Hiper IgE, Wiskott-Aldrich) PTI: púrpura trombocitopénica idiopática IDS: LLC a cél B: con IgG e infecciones. Prevención infecciones bacterianas niños HIV+ Trasplante MO: septicemias, neumonías intersticiales y GVHD. Síndrome de Kawasaki: prevención aneurisma coronario Prevención sepsis neonatal. Aprobadas FDA

59
GGEV: Indicaciones Enfermedad de Kawasaki
Transplante médula ósea adulto Polineuropatía desmielinizante inflamatoria crónica Sindrome de Guillain-Barre. Mieloma múltiple estable con riesgo de infecciones Sindromes vasculíticos sistémicos Anemia hemolítica autoinmune Púrpura trombocitopénica autoinmune Neutropenia mediada por mecanismos inmunes Miastenia gravis descompensada Dermatomiositis y polimiositis 59

60
GGEV GAMMAGLOBULINA EV IgG monomérica < 1% IgA-IgM
400 – 500 mg/kg/mes Mantener niveles de 600mg% de IgG

61
Qué tipo de preparación y qué ruta?
Calidad de inmunoglobulinas disponibles Contenido de IgA Cantidad de sal, azúcar, productos sanguíneos no Igs, agentes estabilizadores, conservantes, pH y osmolaridad. CAUSA DE REACCIONES ADVERSAS!!!

62
Efectos adversos GGEV Efectos menores Cefalea Nauseas Malestar
Mialgias Artralgias Escalofríos Ansiedad Sudoración Calambres intestinales Rash Fiebre en bajo grado Leucopenia Efectos serios Falla renal aguda Accidente cerebrovascular Infarto de miocardio Trombosis venosa profunda Tromboembolismo pulmonar Anafilaxia Meningitis aséptica

63
GGEV REACCIONES ADVERSAS Reacción tipo anafiláctica (Ac anti-IgA)
(hidrocortisona IV (10 mg/ Kg) y difenhidramina IV (1 mg /Kg) Reacción flogística vasodilatadora (1 y el 15%): ansiedad, fiebre y escalofríos, eritema cutáneo, cefalea, mialgias, dolor lumbar o abdominal y vómito. Se las relaciona a altas velocidades de infusión. Cefaleas : días posteriores a infusión. 63
 y difenhidramina IV (1 mg /Kg) Reacción flogística vasodilatadora (1 y el 15%): ansiedad, fiebre y escalofríos, eritema cutáneo, cefalea, mialgias, dolor lumbar o abdominal y vómito. Se las relaciona a altas velocidades de infusión. Cefaleas : días posteriores a infusión. 63.")
64
Velocidad de infusión Inicio: 60-80 ml/h Aumento de 30 ml/h cada 30m
Máximo 200 ml/h

65
Dra Liliana Castro Zorrilla

66
GAMMAGLOBULINAS ENDOVENOSAS
BAXTER BIOGAM Alemania GRIFOLS España PURISSIMUS UNC Kiovig c/g: 1153 $ Citax F c/ g: 968 $ Flebogamma 5%: c/g: 796$ IgG c/ g: 495$ Inmunoglobulina G endovenosa c/ g: 411$

67
GAMMAGLOBULINA CSL Behring Beriglobina® P
Inmunoglobulina humana normal pasteurizada Privigen® 100 mg/ml (al 10%) solución para perfusión Vivaglobin® 160 mg/ml Inmunoglobulina humana normal (subcutánea). CSL Behring
 solución para perfusión. Vivaglobin® 160 mg/ml. Inmunoglobulina humana normal (subcutánea). CSL Behring.")
71
Procesos involucrados en la cobertura de GGEV
Para alcanzar la autosuficiencia en hemoderivados es necesario contar con un nivel de 55 donantes/1000 habitantes/ año. Esta cifra conocida mundialmente debe ser interpretada por la comunidad en general y la comunidad científica como una meta a alcanzar. Se debe recordar que de cada litro de plasma se pueden obtener de 2 a 3 gramos de gammaglobulina, a partir de lo cual se puede calcular fácilmente que cantidad de donaciones necesarias para cada tratamiento.

72
Gammaglobulina Dado que pertenecemos a un sistema de delicado equilibrio, en donde la indicación inadecuada para un paciente perjudica inexorablemente la indicación de otro, todos los esfuerzos deben estar orientados a la optimización de este recurso tan valioso . LEY N° Ciudad Autónoma de Buenos Aires Sanción: 12/08/2004 Promulgación: Decreto Nº 1706 del 13/09/2004 Publicación: BOCBA N° 2028 del 20/09/2004

73
CASOS CLINICOS

74
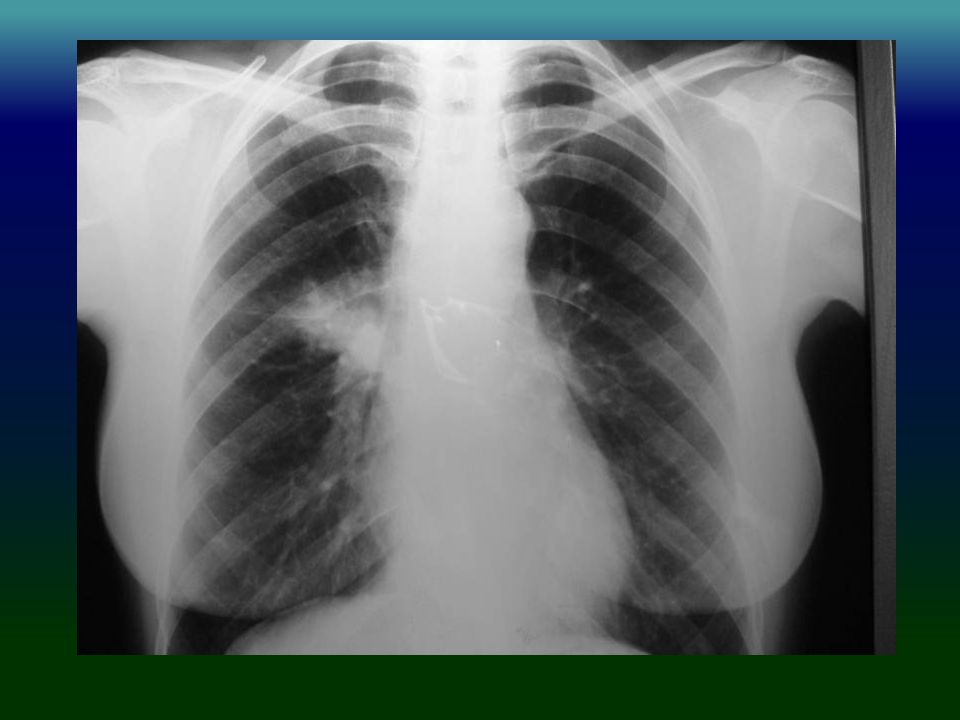
Caso 1: MP, sexo femenino, 42 años.
Consulta por infecciones vías respiratorias altas y bajas a repetición. Bronquiectasias localizadas. Cinco años previos había sufrido lobectomía LID por bronquiectasias sin aún diagnóstico de IDP. Diarreas y síndrome de malabsorción. Dosajes de inmunoglobulinas fueron: IgG: 163 mg %, IgA 19 mg % e IgM: 12 mg %. IgG pos tratamiento: 875 mg %. CD19: 10%. Lleva 10 años de tratamiento sin complicaciones. Diagnóstico: Inmunodeficiencia común variable VN: IgG: IgA: –IgM:

75
Caso 2: AL, sexo femenino, 40 años.
Consulta por infecciones vías respiratorias altas y bajas a repetición. Antecedente de carcinoma folicular tiroideo y gástrico. Los dosajes de inmunoglobulinas fueron: IgG: 400 mg %, IgA ND mg % e IgM: ND mg %. CD19: 8 %. IgG pos tratamiento: 900 mg %. Lleva 15 años de tratamiento con gammaglobulina parenteral sin complicaciones . Diagnóstico: Inmunodeficiencia común variable

76
Caso 3: ML sexo femenino, 30 años.
Consulta por infecciones vías respiratorias altas y bajas a repetición. Diarreas y síndrome de malabsorción. Recibe durante varios meses gammaglobulina intramuscular sin resultados. Los dosajes de inmunoglobulinas: IgG: 100 mg %, IgA ND mg % e IgM: ND mg %. CD 19: 8%. Presenta bronquiectasias bilaterales e insuficiencia respiratoria durante evolución, por lo que debe realizarse oxigenoterapia crónica. IgG pos tratamiento: 790 mg%. Una hermana consulta por granulomatosis pulmonar con diagnóstico de sarcoidosis. Fallece a los 7 años posteriores al tratamiento por insuficiencia respiratoria crónica. Diagnóstico: Inmunodeficiencia común variable

77
Sexo femenino, 30 años, neumonías a repetición.
Diarreas, sindrome malabsorción. Giardias Hermana con granulomatosis pulmonar Sarcoido-like

78
Caso 4: AV, paciente sexo masculino, 50 años
Consulta por infecciones vías respiratorias altas y bajas a repetición. Diarreas y síndrome de malabsorción. Bronquiectasias bilaterales. Tuberculosis pulmonar sin confirmación bacteriológica, recibe tratamiento empírico para tuberculosis. Los dosajes de inmunoglobulinas fueron: IgG: ND mg %, IgA ND mg % e IgM: ND mg %. CD 19: 5%. IgG pos tratamiento: 650 mg %. Realizó 17 años de tratamiento con gammaglobulina Fallece por neumonía e insuficiencia respiratoria Diagnóstico: Inmunodeficiencia común variable

79
Caso 5: VAG, paciente sexo masculino de 30 años
Consulta por infecciones vías respiratorias altas leves y dermatitis eccematosa. Hermano con diagnóstico de IDCV y dos hijos varones con déficit de IgA selectivo. Los dosajes de inmunoglobulinas fueron: IgG: 336 mg %, IgA 58 mg % e IgM: 65 mg%. CD 19: 7%. Anticuerpos antipolisacáridos negativos IgG pos tratamiento: 1000 mg %.. Lleva 11 años de tratamiento sin complicaciones. Diagnóstico: Inmunodeficiencia común variable

80
Caso 6: LR, paciente sexo masculino, 20 años.
Consulta por endoftalmitis y neutropenia. Antecedentes de la infancia: vitiligo desde los 5 años, alopecia areata, sinusitis, bronquitis, diarrea y síndrome de malabsorción. A los 15 años padece una anemia hemolítica autoinmune. Los dosajes de inmunoglobulinas fueron: IgG: ND mg %, IgA 10 mg % e IgM: ND mg %. CD 19: 3%. Isohemaglutininas negativas. IgG pos tratamiento: 800 mg%. Durante la evolución padece cuadros a repetición de AHA y púrpura trombocitopénica idiopática (PTI)
")
81
Posteriormente se diagnosticó una enfermedad inflamatoria intestinal crónica semejante a enfermedad de Crohn y fallece por neumonía e insuficiencia respiratoria aguda a los 10 años de inicio del tratamiento con gammaglobulina. Fue considerado un síndrome poliautoinmune Diagnóstico: Inmunodeficiencia común variable

82
Caso 7: DR, paciente sexo masculino de 18 años
Consulta por infecciones vías respiratorias altas y bajas a repetición. Diarreas y síndrome de malabsorción. Bronquiectasias bibasales. Los dosajes de inmunoglobulinas fueron: IgG: 120 mg %, IgA ND mg % e IgM: ND mg %. CD 19: 0 %. IgG pos tratamiento: 550 mg %. En el interrogatorio: fallecimiento de hermano sexo masculino por infecciones respiratorias recidivantes. Otro hermano también de sexo masculino a quien se le diagnostica una mutación del BTK. Realiza 4 años de tratamiento con internaciones por neumonías en 2 oportunidades. Fallece por neumonía asociada a Virus Gripe A Diagnóstico: Agmmaglobulinemia LX o Enfermedad de Bruton

84
Caso 8: AG, 18 años , sexo masculino.
Antecedentes de otitis reiteradas desde nacimiento, con pérdida oido izquierdo. Neumonía a los 11 años. Consulta por diarrea y sind malabsorción, en estudio por enf celíaca y fibrosis quística. Esplenomegalia. Isohemaglutininas neg. Dosajes de Ig A G M no dosables. Comienza gammaglobulina parenteral hace 2 meses y recupera 10 kilos en 40 días Diagnóstico: Inmunodeficiencia común variable

85
Caso 9: NR 57 años, sexo femenino
2010 neumonía. Tos catarro a repetición, asma bronquial . Dosajes Igs: A 136, G 370 y M 155 Isohemag bajas. Hiperglucemia Diabetes que requiere insulina Diagnóstico: Inmunodeficiencia común variable

86
Caso 10: DVC , 36 años, sexo masculino
asma bronquial . Derivado por un hallazgo en proteinograma con ausencia banda gammaglobulinas Dosajes Igs: A 20, G 375 y M 20 Neumonía bacteriana y adenopatías mediastinales y axilares compatibles con linfoma Diagnóstico: Inmunodeficiencia común variable

87
Caso 11: NA, sexo femenino, 51 años.
Antecedentes: sinusitis a repetición, bronquitis y neumonía LMD. Bronquiectasias. Hipotiroidismo. Dosajes IgA ND, IgG 711, IgM: 31 Continúa con infecciones respiratorias. 4 años posteriores al Diagnóstico de Inmunodef Selectiva de IgA, en TAC TX de control, se observan múltiples adenomegalias mediastino superior y peritraqueales: Biopsia compatible con Sarcoidosis. Ultimo control Igs: IgA ND, IgG 491, IgM 24, con falta de respuesta a anticuerpos antipolisacáridos y toxoide tetánico: Diagnóstico Inmunodeficiencia común variable.

90
Caso 12: NP paciente sexo femenino
Consulta a los 20 años por infecciones vías respiratorias altas y bajas a repetición. Diarreas y síndrome de malabsorción. En la fecha del diagnóstico los dosajes de inmunoglobulinas fueron: IgG: 100 mg %, IgA 54 mg % e IgM: 85 mg %. CD 19: 6%. IgG pos tratamiento: 500 mg %. Presenta bronquiectasias bilaterales. Desarrolla una encefalitis viral crónica con demencia secundaria. Fallece a los 15 años posteriores al tratamiento.

92
INMUNODEFICIENCIAS PRIMARIAS CONCLUSIONES
Déficit Humoral NO sólo en pediatría Complicaciones más frecuentes : Infecciones recurrentes Vías Respiratorias Bronquiectasias EPOC Causa de Muerte: Insuficiencia Respiratoria

93
The Persistence of Memory, 1931, Salvador Dali

Presentaciones similares