Descargar la presentación
La descarga está en progreso. Por favor, espere

1
Velocidad de las reacciones quimicas
Tema 10 2013

2
Introducción Por medio de la termodinámica es posible predecir la ocurrencia de reacciones, pero no se habla de un aspecto que es de suma importancia y que tiene relación con el tiempo en el cual se llevan a cabo estos procesos. Precisando la inestabilidad relativa de los estados inicial y final de la reacción, la termodinámica permite determinar no sólo si el sistema es susceptible de evolucionar, sino también cual es el límite de esta evolución. La termodinámica permite decidir si los intentos para llevar a cabo una determinada reacción están justificados o no.

3
En su aplicación a las reacciones reales, la termodinámica química requiere el conocimiento de datos termodinámicos relativos a las sustancias reaccionantes y a aquellas susceptibles de ser obtenidas como productos de la reacción. El principio motor de una reacción puede ser resumido en dos importantes criterios: El primer criterio ya fue establecido por Berthelot y según el cual, un sistema químico tiende a evolucionar de tal manera que libere la mayor cantidad posible de calor. El segundo criterio constitutivo del principio motor de los procesos químicos es el criterio de máximo incremento de entropía.

4
Los dos criterios anteriores, constitutivos del principio motor, pueden influir en el mismo sentido o en sentidos opuestos. En el primer caso, la reacción será, desde el punto de vista termodinámico, fácil y completa si los dos criterios actúan en sentido favorable, o bien será imposible si ambos criterios actúan en sentido desfavorable. En el segundo caso, cuando los dos criterios se contrarrestan, se llega a una situación de equilibrio más o menos avanzada, según su importancia relativa.

5
De acuerdo con nuestros conocimientos termodinámicos actuales, sabemos que:
Un valor negativo de y uno positivo de es lo ideal para que K tenga un valor grande y, por tanto, la reacción sea completa. En cambio, un valor positivo de y uno negativo de conlleva un valor muy pequeño de K y la reacción resulta irrealizable. (1)
")
6
Podemos considerar gráficamente las distintas posibilidades que pueden presentarse.

7
De acuerdo con el anterior esquema, una reacción exotérmica acompañada de un aumento de entropía será prácticamente completa, mientras que otra endotérmica con disminución de entropía será irrealizable. Cuando los incrementos de entalpía y de entropía son del mismo signo (positivos o negativos ambos) dependerá de su importancia relativa para que la situación de equilibrio que se alcance esté más o menos desplazada hacia la formación de productos.
 dependerá de su importancia relativa para que la situación de equilibrio que se alcance esté más o menos desplazada hacia la formación de productos..")
8
La termodinámica química permite determinar si una reacción es posible, en qué condiciones experimentales conviene realizarla y, fijadas éstas, cual será el grado de conversión máximo que se puede esperar. También nos da la cantidad de calor, y eventualmente la de trabajo mecánico, que el sistema suministrará o deberá absorber para conseguir la transformación deseada. Por el contrario, y éstas son sus limitaciones, la termodinámica química no da información acerca del “camino” que el sistema reactivo debe recorrer para pasar de los reactivos a los productos (es decir, de su “mecanismo”) ni de la “rapidez” con la que se producirá el proceso químico (el tiempo no es una variable que aparezca en la termodinámica del equilibrio).
 ni de la rapidez con la que se producirá el proceso químico (el tiempo no es una variable que aparezca en la termodinámica del equilibrio).")
9
Asimismo, cuando la transformación química implica una cascada de reacciones consecutivas, la termodinámica no indica tampoco en qué etapa se detendrá ni en qué proporción se realizará un determinado proceso cuando se presenten simultáneamente varias opciones de reacción. Las limitaciones de la termodinámica, son precisamente los objetivos esenciales de la cinética química. Cuando una reacción ya ha sido estudiada desde el punto de vista termodinámico, y se ha comprobado su viabilidad, la siguiente etapa consiste en su estudio cinético.

10
El objetivo de la cinética química es el estudio de las velocidades de las reacciones químicas y de los factores de los que dependen dichas velocidades. De estos factores, los más importantes son la concentración y la temperatura. Haciendo un estudio sistemático de los efectos de estos factores sobre las velocidades, se pueden sacar conclusiones sobre el mecanismo por el que se verifican las reacciones químicas.

11
La cinética pretende dar respuesta a los siguientes problemas:
a) Encontrar la forma adecuada para activar la reacción. La necesidad de activar las reacciones químicas aparece, en primer lugar, por la existencia de numerosos sistemas metaestables aparentemente estables, pero capaces de evolucionar con el estímulo adecuado. Podemos considerar distintas formas de activar una reacción. Por ejemplo
 Encontrar la forma adecuada para activar la reacción. La necesidad de activar las reacciones químicas aparece, en primer lugar, por la existencia de numerosos sistemas metaestables aparentemente estables, pero capaces de evolucionar con el estímulo adecuado. Podemos considerar distintas formas de activar una reacción. Por ejemplo.")
12
a.1. Activación térmica. La elevación de la temperatura tiene como resultado la exaltación de la agitación térmica intermolecular e intramolecular, lo cual favorece la ruptura y formación de enlaces. a.2. Activación catalítica. Las limitaciones y poca selectividad de la activación térmica conducen a investigar medios de activación más eficaces. Uno de ellos es el empleo de un catalizador. Los catalizadores no se consumen en la reacción y su labor no implica ninguna aportación de energía al sistema químico.

13
a. 3. Activación fotoquímica
a.3. Activación fotoquímica. Ciertos sistemas químicos son capaces de reaccionar cuando se les somete a la acción de la luz. Esto permite activar uno de los reactivos, el causante de la absorción luminosa. a.4. Activación por inducción. En este caso una reacción auxiliar puede proporcionar la energía necesaria para que un sistema evolucione. a.5. Activación eléctrica. Bajo esta expresión se agrupan dos tipos de fenómenos distintos, según si la corriente eléctrica actúa en un medio ionizado (este es el caso de los procesos electrolíticos) o simplemente se hace saltar una chispa o descarga eléctrica para iniciar una combustión.
 o simplemente se hace saltar una chispa o descarga eléctrica para iniciar una combustión.")
14
b) Delimitación de la fase o interfase donde se produce la reacción
b) Delimitación de la fase o interfase donde se produce la reacción. A primera vista esta cuestión puede parecer simple y sin notables consecuencias, pero es el origen de una importante serie de dificultades con la que se encuentra el experimentalista en el laboratorio e incluso en su extrapolación hasta la escala industrial. Una reacción que ha tenido lugar de modo uniforme en una sola fase se denomina homogénea, si no es así, se dice que la reacción es heterogénea.
 Delimitación de la fase o interfase donde se produce la reacción. A primera vista esta cuestión puede parecer simple y sin notables consecuencias, pero es el origen de una importante serie de dificultades con la que se encuentra el experimentalista en el laboratorio e incluso en su extrapolación hasta la escala industrial. Una reacción que ha tenido lugar de modo uniforme en una sola fase se denomina homogénea, si no es así, se dice que la reacción es heterogénea.")
15
c) Identificación de las formas activas
c) Identificación de las formas activas. La ruptura y la formación de enlaces que acompaña a toda reacción química se realizan a través de una gran variedad de procesos en los que intervienen diferentes formas intermedias activas. La naturaleza de las formas activas de la reacción determina la elección del medio más propicio para su formación (medio polar como es el medio acuoso o un medio apolar).
 Identificación de las formas activas. La ruptura y la formación de enlaces que acompaña a toda reacción química se realizan a través de una gran variedad de procesos en los que intervienen diferentes formas intermedias activas. La naturaleza de las formas activas de la reacción determina la elección del medio más propicio para su formación (medio polar como es el medio acuoso o un medio apolar).")
16
Velocidad de reacción, ecuación de velocidad
A la hora de definir la velocidad de reacción, la primera cosa que hay que tener clara es su estequiometría, ya que la rapidez con que se consumen los reactivos y se forman los productos se encuentran relacionadas a través de los coeficientes estequiométricos. Para una reacción química general, donde (2)
")
17
El valor de x es único para cada reacción
Los moles de reactivos y productos están relacionados con el grado de conversión según donde es el número de moles iniciales de la especie química i (reactivo o producto) y es el grado de avance de la reacción (magnitud extensiva). Para la ecuación gral: (3) El valor de x es único para cada reacción
 y es el grado de avance de la reacción (magnitud extensiva). Para la ecuación gral: (3) El valor de x es único para cada reacción.")
18
Derivando (3) respecto del tiempo se tiene
A la cantidad se le denomina velocidad de conversión (J) y es, al igual que , una magnitud extensiva. Si representamos por J dicha velocidad, a partir de su definición y de (4), se tiene (4)
 y es, al igual que , una magnitud extensiva. Si representamos por J dicha velocidad, a partir de su definición y de (4), se tiene. (4)")
19
(5) De (5) se infiere que J, definida de esta forma, siempre es una cantidad positiva y es independiente de la especie química i (reactivo o producto) a la cual esté referida. De acuerdo con la definición de concentración molar: llevando esta última expresión a (5) se tiene (6)
 se infiere que J, definida de esta forma, siempre es una cantidad positiva y es independiente de la especie química i (reactivo o producto) a la cual esté referida. De acuerdo con la definición de concentración molar: llevando esta última expresión a (5) se tiene. (6)")
20
Dividiendo la expresión (6) por el volumen obtenemos una velocidad de conversión específica (velocidad por unidad de volumen) que es lo que consideramos velocidad de reacción v: (7) Si el volumen del sistema reactivo permanece constante, la velocidad de reacción queda reducida a, Para la reacción química general, Unidades: mol m-3 s-1 mol L s-1 molécula cm-3 s-1 (8)
 Si el volumen del sistema reactivo permanece constante, la velocidad de reacción queda reducida a, Para la reacción química general, Unidades: mol m-3 s-1. mol L s-1. molécula cm-3 s-1. (8)")
21
Ecuación de velocidad empírica. Orden de reacción.
En ciertos casos (cinéticas sencillas) la ley de velocidad tiene la forma (9) a, b, ... : órdenes parciales de la reacción k: constante conocida como constante de velocidad Unidades: s-1 m3 mol-1 s-1 L mol-1 s-1 cm3 molécula-1 s-1 Orden Total de Reacción: n = a + b +
 la ley de velocidad tiene la forma. (9) a, b, ... : órdenes parciales de la reacción. k: constante conocida como constante de velocidad. Unidades: s-1. m3 mol-1 s-1. L mol-1 s-1. cm3 molécula-1 s-1. Orden Total de Reacción: n = a + b + ")
22
Las reacciones químicas elementales, es decir, aquellas que transcurren en un solo acto, tienen (siempre) leyes de velocidad del tipo (9). En estos casos los órdenes de la reacción respecto de los reactivos implicados coinciden con los coeficientes estequiométricos y el orden global con la molecularidad del proceso. Así, por ejemplo, la reacción entre el hidrógeno y el yodo (la cual, es una reacción que transcurre en un solo paso) es una reacción elemental bimolecular, con lo que
 es una reacción elemental bimolecular, con lo que.")
23
En cambio, la reacción entre el H2 y el Br2 para dar bromuro de hidrógeno:
en fase gaseosa y a temperaturas comprendidas entre 200 ºC y 300 ºC, fue estudiada por Bodenstein y Lind en 1906, demostrando que la reacción no era elemental, sino más bien, transcurría mediante el siguiente mecanismo en cadena:

25
La etapa 1 es la de iniciación, las etapas 2 y 3 son las de propagación (la suma de éstas da la reacción global), la etapa 4 es la inversa de la 2 y se trata de una inhibición, finalmente, la etapa 5 es la de terminación de la cadena. Puede observarse que en las 5 etapas del mecanismo anterior las leyes de velocidad de cada una de ellas se ajustan a la expresión (9), coincidiendo los órdenes parciales con los coeficientes estequiométricos y siendo el orden global igual a la molecularidad de la etapa. Bodenstein y Lind obtuvieron, experimentalmente, para la reacción, la siguiente ley de velocidad

26
que, como fácilmente puede concluirse, no se ajusta a la cinética sencilla dada por la expresión (9).
.")
27
Concepto de reacción, mecanismo y molecularidad
Por reacción entendemos el proceso químico, simple o complejo en su detalle, por el cual los reactivos constituyentes de un determinado sistema reactivo se transforman, en una proporción fija y definida, en un sistema de productos relacionados estequiométricamente. Podemos clasificar las reacciones en la siguiente forma: a) Reacciones aisladas. Transformación química en la que, aparte de las formas fugaces que tan solo son detectables mediante técnicas especiales, únicamente aparece un único sistema de productos. Son del tipo
 Reacciones aisladas. Transformación química en la que, aparte de las formas fugaces que tan solo son detectables mediante técnicas especiales, únicamente aparece un único sistema de productos. Son del tipo.")
28
b) Reacciones compuestas
b) Reacciones compuestas. Son aquellas transformaciones en las que transitoriamente, o al final de la reacción, aparecen varios sistemas de productos. Podemos distinguir los siguientes casos b1) reacciones reversibles (también llamadas opuestas o equilibradas). La transformación viene limitada por un equilibrio químico que constituye el estado final de la misma. Este estado de equilibrio, que puede ser alcanzado ya sea partiendo de los reactivos como de los productos, resulta de la igualdad de las velocidades de las dos reacciones opuestas. Podemos simbolizarlas como Como puede observarse, tenemos dos sistemas de productos: C+D como productos de la reacción directa y A+B como productos de la reacción inversa.
 Reacciones compuestas. Son aquellas transformaciones en las que transitoriamente, o al final de la reacción, aparecen varios sistemas de productos. Podemos distinguir los siguientes casos. b1) reacciones reversibles (también llamadas opuestas o equilibradas). La transformación viene limitada por un equilibrio químico que constituye el estado final de la misma. Este estado de equilibrio, que puede ser alcanzado ya sea partiendo de los reactivos como de los productos, resulta de la igualdad de las velocidades de las dos reacciones opuestas. Podemos simbolizarlas como. Como puede observarse, tenemos dos sistemas de productos: C+D como productos de la reacción directa y A+B como productos de la reacción inversa.")
29
b2) Reacciones paralelas irreversibles
b2) Reacciones paralelas irreversibles. Son aquellas que se transforman simultáneamente en varias direcciones. Podemos distinguir b2.1) Reacciones gemelas. Si tienen en común los mismos reactivos b2.2) Reacciones concurrentes o competitivas. Cuando los reactivos no son todos comunes, estableciéndose, por tanto, una competencia entre los reactivos no comunes para la transformación de los reactivos comunes. Simbólicamente:
 Reacciones paralelas irreversibles. Son aquellas que se transforman simultáneamente en varias direcciones. Podemos distinguir. b2.1) Reacciones gemelas. Si tienen en común los mismos reactivos. b2.2) Reacciones concurrentes o competitivas. Cuando los reactivos no son todos comunes, estableciéndose, por tanto, una competencia entre los reactivos no comunes para la transformación de los reactivos comunes. Simbólicamente:")
30
b3) Reacciones consecutivas
b3) Reacciones consecutivas. Conjunto de transformaciones químicas tal que el producto de una etapa es el reactivo de la etapa siguiente c) Reacción supercompuesta. Cuando en la transformación química intervienen simultáneamente dos o más reacciones del tipo b. Por mecanismo de una reacción entendemos el proceso detallado (o conjunto de reacciones elementales) por medio del cual los reactivos se transforman en productos. El establecimiento o determinación del mecanismo de una cierta reacción suele ser un proceso experimental complicado y exige cierto grado de ingenio para la interpretación de los datos cinéticos obtenidos.
 Reacciones consecutivas. Conjunto de transformaciones químicas tal que el producto de una etapa es el reactivo de la etapa siguiente. c) Reacción supercompuesta. Cuando en la transformación química intervienen simultáneamente dos o más reacciones del tipo b. Por mecanismo de una reacción entendemos el proceso detallado (o conjunto de reacciones elementales) por medio del cual los reactivos se transforman en productos. El establecimiento o determinación del mecanismo de una cierta reacción suele ser un proceso experimental complicado y exige cierto grado de ingenio para la interpretación de los datos cinéticos obtenidos.")
31
A menudo puede ocurrir que nos encontremos con más de un mecanismo compatible con los datos cinéticos obtenidos por vía experimental. En estas circunstancias el análisis cinético es insuficiente para inclinarnos por una u otra opción. El mecanismo de reacción nunca es obtenible a partir de la simple inspección de la estequiometría del proceso. Así, podemos encontrarnos con situaciones tan diferentes como a) Reacción entre el H2 y el I2 para dar HI:
 Reacción entre el H2 y el I2 para dar HI:")
32
En este caso el mecanismo consta de una sola etapa que coincide con la reacción global o estequiométrica especificada anteriormente. Se trata de una reacción elemental, es decir una reacción que tiene lugar en un solo acto. En ella una molécula de H2 “choca” contra una de I2 formando un aducto o complejo intermedio de vida muy efímera y energía alta, a partir del cual se forman los productos. Esquemáticamente

33
b) Descomposición térmica del acetaldehído para dar CH4 y CO:
Cada molécula de acetaldehído no se rompe en un paso sencillo para dar una molécula de metano y una de monóxido de carbono. El mecanismo es el siguiente: Cada una de las tres etapas del mecanismo anterior constituye una reacción elemental porque transcurre en un solo acto.

34
Un concepto asociado única y exclusivamente a las reacciones elementales es el concepto de molecularidad. La molecularidad de una reacción química elemental (aquella que se produce en un solo acto) se define como el número de moléculas de reactivo que intervienen en ella. Dado que una reacción química elemental debe producirse por el “choque” de las especies químicas reactivas, es poco probable que en un instante determinado se encuentren más de tres moléculas; por ello las reacciones elementales pueden ser monomoleculares, bimoleculares y trimoleculares. Son muy raros los casos en que la molecularidad es 4. Reacción elemental monomolecular. Una reacción de este tipo implica una sola molécula de reactivo y es una isomerización ( A →B) o una descomposición (A →B+C).
 o una descomposición (A →B+C).")
35
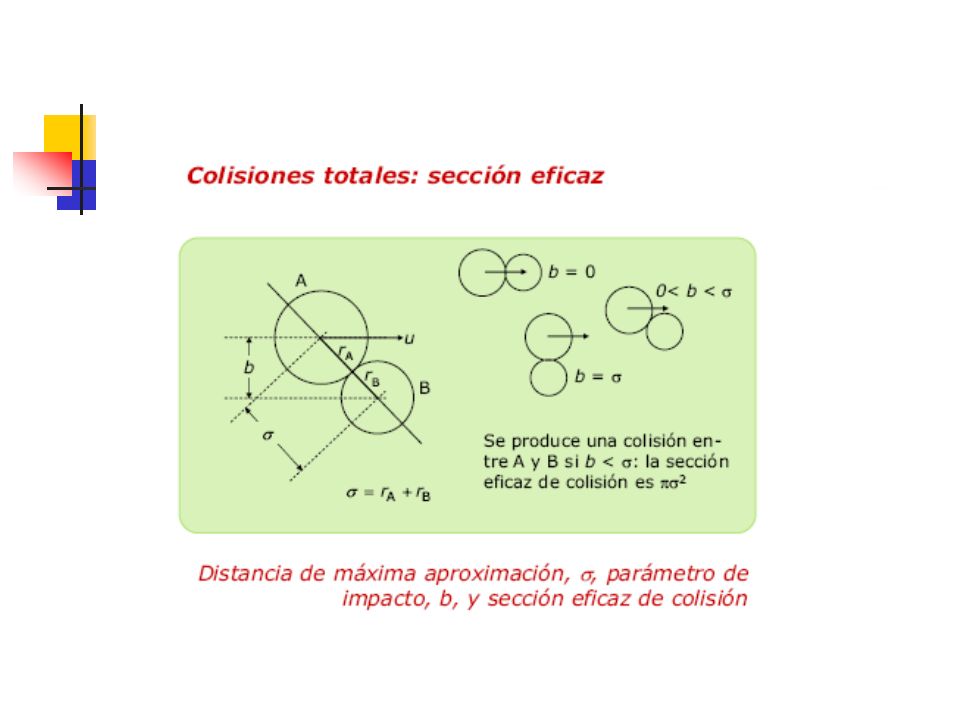
Reacción elemental bimolecular
Reacción elemental bimolecular. En estas reacciones dos moléculas de reactivo, iguales o diferentes, se combinan para dar un solo producto o un número de moléculas de producto. O bien son reacciones de asociación (A+B →AB, 2A→A2) o bien reacciones de canje (A+B → C+D, 2A→C+D). Reacción elemental trimolecular. No son muy frecuentes ya que implican la colisión simultanea de tres moléculas para dar uno o varios productos (A+B+C→ productos).
 o bien reacciones de canje (A+B → C+D, 2A→C+D). Reacción elemental trimolecular. No son muy frecuentes ya que implican la colisión simultanea de tres moléculas para dar uno o varios productos (A+B+C→ productos).")
36
Para terminar el mecanismo de una reacción vamos a ver una serie de criterios que debe cumplir cualquier mecanismo de reacción: a) Consistencia con los resultados experimentales. Es fácil proponer un mecanismo para una reacción cuando se dispone de muy poca información acerca de la misma. En tales casos es difícil probar o desechar dicho mecanismo. Sin embargo, a medida que se van obteniendo más datos experimentales, resulta más difícil encontrar un mecanismo que satisfaga todos los resultados conocidos. b) Viabilidad energética. Cuando ocurre una reacción de descomposición, es el enlace más débil de la molécula el que se rompe. Así en la descomposición del peróxido de dibutilo terciario, (CH3)3COOC(CH3)3, es el enlace O-O el que se rompe inicialmente dando dos radicales butoxilo terciario, (CH3)3CO.
 Consistencia con los resultados experimentales. Es fácil proponer un mecanismo para una reacción cuando se dispone de muy poca información acerca de la misma. En tales casos es difícil probar o desechar dicho mecanismo. Sin embargo, a medida que se van obteniendo más datos experimentales, resulta más difícil encontrar un mecanismo que satisfaga todos los resultados conocidos. b) Viabilidad energética. Cuando ocurre una reacción de descomposición, es el enlace más débil de la molécula el que se rompe. Así en la descomposición del peróxido de dibutilo terciario, (CH3)3COOC(CH3)3, es el enlace O-O el que se rompe inicialmente dando dos radicales butoxilo terciario, (CH3)3CO.")
37
En un mecanismo en el que están implicados átomos o radicales libres, aquel proceso que sea exotérmico, o el menos endotérmico, es muy probable que sea un paso importante en la reacción. c) Principio de reversibilidad microscópica. Este principio establece que en el caso de una reacción elemental, la reacción inversa ocurre por el mismo camino pero en sentido opuesto. En consecuencia no es posible incluir en un mecanismo de reacción ningún paso que no pueda tener lugar si la reacción se invirtiera. Por ejemplo, en la descomposición térmica del peróxido de dibutilo terciario, no es posible postular el paso inicial como:
 Principio de reversibilidad microscópica. Este principio establece que en el caso de una reacción elemental, la reacción inversa ocurre por el mismo camino pero en sentido opuesto. En consecuencia no es posible incluir en un mecanismo de reacción ningún paso que no pueda tener lugar si la reacción se invirtiera. Por ejemplo, en la descomposición térmica del peróxido de dibutilo terciario, no es posible postular el paso inicial como:")
38
(CH3)3COOC(CH3)3 → 6 CH3 • + 2 CO dado que el paso inverso no puede tener lugar (se trataría de una reacción elemental con molecularidad 8). Dado que las etapas elementales de un mecanismo de reacción son monomoleculares, bimoleculares o a lo sumo trimoleculares, cualquier mecanismo propuesto no debe contener etapas (elementales) que den lugar a más de tres moléculas de producto.
. Dado que las etapas elementales de un mecanismo de reacción son monomoleculares, bimoleculares o a lo sumo trimoleculares, cualquier mecanismo propuesto no debe contener etapas (elementales) que den lugar a más de tres moléculas de producto.")
39
ANÁLISIS DE DATOS EXPERIMENTALES.
Vamos a considerar únicamente aquellas reacciones que presentan una cinética sencilla cuyas leyes de velocidad son del tipo indicado en la expresión (9). Para cada uno de los casos que consideremos veremos de qué forma los datos experimentales confirman la idoneidad de la ley de velocidad supuesta (es decir, haremos lo que se denomina un análisis de datos cinéticos). En toda investigación cinética se mide, de forma directa o indirecta, la concentración a diversos tiempos.
. Para cada uno de los casos que consideremos veremos de qué forma los datos experimentales confirman la idoneidad de la ley de velocidad supuesta (es decir, haremos lo que se denomina un análisis de datos cinéticos). En toda investigación cinética se mide, de forma directa o indirecta, la concentración a diversos tiempos.")
40
el problema es encontrar la ley de velocidad, es decir, una ecuación que relacione la velocidad de reacción con la concentración de los reactivos, y a veces de los productos y otras sustancias presentes (por ejemplo catalizadores). Cuando la reacción responde a un orden (como es el caso que vamos a considerar) debemos determinar cual es y también la constante de velocidad (constante k en la expresión (9)).
 debemos determinar cual es y también la constante de velocidad (constante k en la expresión (9))..")
41
Los métodos para obtener los datos experimentales son básicamente de dos tipos:
1.a) Métodos de extracción de muestras. Estos métodos implican la extracción de una muestra de la mezcla reaccionante con objeto de proceder a su análisis. La muestra extraída (del recipiente en el que está teniendo lugar la reacción) se enfría o se diluye para detener la reacción y de esta forma poder medir la concentración del reactivo adecuado. El procedimiento se repite a diferentes tiempos de reacción. Para analizar las muestras extraídas se suelen utilizar fundamentalmente técnicas de valoración, de cromatografía de gases o de espectrometría de masas.
 Métodos de extracción de muestras. Estos métodos implican la extracción de una muestra de la mezcla reaccionante con objeto de proceder a su análisis. La muestra extraída (del recipiente en el que está teniendo lugar la reacción) se enfría o se diluye para detener la reacción y de esta forma poder medir la concentración del reactivo adecuado. El procedimiento se repite a diferentes tiempos de reacción. Para analizar las muestras extraídas se suelen utilizar fundamentalmente técnicas de valoración, de cromatografía de gases o de espectrometría de masas.")
42
1.b) Métodos continuos. Existe una serie de métodos cinéticos en los que se mide una propiedad física del sistema reaccionante a distintos intervalos de tiempo durante el transcurso de la reacción. De esta manera la mezcla reaccionante no se perturba por la recogida de muestras y se permite que la reacción transcurra hasta su terminación. Algunos métodos continuos son el método de la conductividad eléctrica, el método de la rotación óptica, el método espectrofotométrico, etc.

43
Para determinar el orden y la constante de velocidad (encontrar la expresión de la velocidad) hay diversos métodos, los principales son: 1) Método de integración. 2) Método diferencial 3) Método de aislamiento 1- En el método de integración se parte de una ecuación cinética que se supone aplicable; por ejemplo, si se cree que la reacción es de primer orden se parte de
 Método de integración. 2) Método diferencial. 3) Método de aislamiento. 1- En el método de integración se parte de una ecuación cinética que se supone aplicable; por ejemplo, si se cree que la reacción es de primer orden se parte de.")
44
donde A es el coeficiente estequiométrico de la especie química A, [A] es la concentración molar de A y k es la constante de velocidad. Esta ecuación se transforma por integración en otra donde se expresa [A] en función del tiempo t. Si la ecuación obtenida por integración presenta una buena concordancia con los datos experimentales de concentración frente a tiempo, entonces podemos afirmar que el orden es efectivamente uno, y mediante un procedimiento gráfico se puede calcular la constante de velocidad k.
![donde A es el coeficiente estequiométrico de la especie química A, [A] es la concentración molar de A y k es la constante de velocidad. Esta ecuación se transforma por integración en otra donde se expresa [A] en función del tiempo t.](http://slideplayer.es/slide/27139/1/images/44/donde+%EF%81%AEA+es+el+coeficiente+estequiom%C3%A9trico+de+la+especie+qu%C3%ADmica+A%2C+%5BA%5D+es+la+concentraci%C3%B3n+molar+de+A+y+k+es+la+constante+de+velocidad.+Esta+ecuaci%C3%B3n+se+transforma+por+integraci%C3%B3n+en+otra+donde+se+expresa+%5BA%5D+en+funci%C3%B3n+del+tiempo+t..jpg "Si la ecuación obtenida por integración presenta una buena concordancia con los datos experimentales de concentración frente a tiempo, entonces podemos afirmar que el orden es efectivamente uno, y mediante un procedimiento gráfico se puede calcular la constante de velocidad k.")
45
Si no hubiera buena concordancia, hay que buscar otra ecuación cinética y proceder análogamente hasta obtener un resultado satisfactorio. Este método parece ser algo así como “probar fortuna”, pero a pesar de ello es muy valioso, sobre todo cuando no surgen complicaciones especiales.

46
2-El método diferencial emplea la ecuación cinética en su forma diferencial, sin integrar. Es conocido como el método de Van’t Hoff y se aplica a leyes de velocidad del tipo (donde n es el orden de la reacción). Este método se basa en la determinación de las velocidades reales a partir de la pendiente de la curvas concentración − tiempo (obtenidas experimentalmente, Figura 1): Figura 1
. Este método se basa en la determinación de las velocidades reales a partir de la pendiente de la curvas concentración − tiempo (obtenidas experimentalmente, Figura 1): Figura 1.")
47
La principal dificultad de este método estriba en que las pendientes no pueden obtenerse con mucha exactitud; a pesar de esto el método es en conjunto el más seguro y, a diferencia de lo que ocurre con el método integral, no surgen dificultades particulares cuando el comportamiento cinético es complejo. En esencia el método consiste en lo siguiente: A partir de la expresión (10)
")
48
tomando logaritmos tenemos
(11) Por tanto, si se determina la velocidad para distintos valores de la concentración de reactivo, la representación gráfica de ln v frente a ln[A] nos dará una recta cuya pendiente será el orden n y cuya ordenada en el origen será ln k (a partir de la cual obtendremos k). En efecto, de la figura 1 o 2, podemos obtener las pendientes (y por tanto las velocidades) para diferentes puntos ([A], t).
 Por tanto, si se determina la velocidad para distintos valores de la concentración de reactivo, la representación gráfica de ln v frente a ln[A] nos dará una recta cuya pendiente será el orden n y cuya ordenada en el origen será ln k (a partir de la cual obtendremos k). En efecto, de la figura 1 o 2, podemos obtener las pendientes (y por tanto las velocidades) para diferentes puntos ([A], t).")
49
Gráficamente: (Figura 2)
")
50
Este método presenta una variante en la que se centra la atención en las velocidades iniciales. Para el estado inicial de la reacción la ecuación (11) queda en la forma (12) en la que v0 es la velocidad inicial y [A]0 representa la concentración inicial de la especie química A. Por tanto, si se determina la velocidad para distintos valores de la concentración inicial, una representación de ln v0 frente a ln[A]0 dará una recta cuya pendiente será el orden n de la reacción y cuya ordenada en el origen nos permitirá obtener la constante de velocidad k. En esta variante del método diferencial hay que realizar distintas experiencias partiendo de diferentes valores de [A]0. Para cada una de estas experiencias dibujamos una curva [A]-t semejante a la representada en la figura 2.
 en la que v0 es la velocidad inicial y [A]0 representa la concentración inicial de la especie química A. Por tanto, si se determina la velocidad para distintos valores de la concentración inicial, una representación de ln v0 frente a ln[A]0 dará una recta cuya pendiente será el orden n de la reacción y cuya ordenada en el origen nos permitirá obtener la constante de velocidad k. En esta variante del método diferencial hay que realizar distintas experiencias partiendo de diferentes valores de [A]0. Para cada una de estas experiencias dibujamos una curva [A]-t semejante a la representada en la figura 2.")
51
Figura 3 nc

52
Para cada una de las curvas dibujadas debemos obtener la pendiente en el origen (pendiente para t = 0); a partir de la cual se obtiene la velocidad inicial v0. Una vez que tenemos las velocidades iniciales correspondientes a distintas concentraciones iniciales [A]0, representando ln v0 frente a ln[A]0 se obtiene una recta cuya pendiente es n y cuya ordenada en el origen es ln k.

53
Al principio de una reacción se puede tener la certeza de lo que hay presente en el sistema reaccionante; por el contrario en etapas posteriores pueden aparecer productos intermedios que interfieran el curso de la reacción. Por tanto el procedimiento de las velocidades iniciales elimina posibles complicaciones debidas a interferencias entre los productos, y conduce a un orden que corresponde a la situación más sencilla. Así, no es de extrañar que ambas modalidades del método diferencial conduzca, en ocasiones, a órdenes de reacción diferentes.

54
3- El método de aislamiento se utiliza en cinéticas del tipo:
donde el orden global de la reacción será α + β Estas ecuaciones de velocidad no son susceptibles de ser tratadas por los dos métodos anteriores (a no ser que todas las concentraciones iniciales, así como todos los coeficientes estequiométricos de los reactivos, sean iguales; con lo cual la ecuación de velocidad degenera en una del tipo

55
Con el objeto de encontrar los órdenes parciales α, β,
Con el objeto de encontrar los órdenes parciales α, β, ..., el procedimiento más adecuado consiste en definir las condiciones iniciales de los experimentos a realizar utilizando la siguiente estrategia: Todos los reactivos, a excepción de uno, se introducen en gran exceso. De esta forma, las concentraciones de los reactivos en exceso apenas variarán durante el transcurso de la reacción (tener en cuenta que el reactivo en defecto actúa como limitante). Así, si suponemos que todos menos el A se encuentran inicialmente en exceso, la ecuación de velocidad resultará
. Así, si suponemos que todos menos el A se encuentran inicialmente en exceso, la ecuación de velocidad resultará.")
56
es, para esta experiencia, una constante aparente (seudo-constante)
es, para esta experiencia, una constante aparente (seudo-constante). En estas condiciones, cualquier método integral o diferencial nos dará los valores de la seudo-constante k’ y del orden parcial . Si en una segunda experiencia hacemos que todos los reactivos excepto B se encuentren en exceso, un razonamiento análogo nos dará ' k’B y β . Finalmente, con todos los resultados obtenidos será inmediato calcular la verdadera constante de velocidad k.
. En estas condiciones, cualquier método integral o diferencial nos dará los valores de la seudo-constante k’ y del orden parcial . Si en una segunda experiencia hacemos que todos los reactivos excepto B se encuentren en exceso, un razonamiento análogo nos dará k’B y β . Finalmente, con todos los resultados obtenidos será inmediato calcular la verdadera constante de velocidad k.")
57
Método de integración. 1-Cinética de primer orden.
Supongamos que la reacción es de primer orden. De acuerdo con la ecuación (8) y (9) tendremos Como el coeficiente es porque A es reactivo
 y (9) tendremos. Como el coeficiente es porque A es reactivo.")
58
Separando variables e integrando
(13) La ecuación (13) puede ser escrita de la siguiente forma: de donde se infiere que una representación de ln[A] frente al tiempo, si la reacción estudiada es realmente de primer orden, debe dar una recta de pendiente νA k (téngase en cuenta que si A es un reactivo el coeficiente estequiométrico νA es negativo y, por tanto, la pendiente de la recta resulta negativa): (14)
 La ecuación (13) puede ser escrita de la siguiente forma: de donde se infiere que una representación de ln[A] frente al tiempo, si la reacción estudiada es realmente de primer orden, debe dar una recta de pendiente νA k (téngase en cuenta que si A es un reactivo el coeficiente estequiométrico νA es negativo y, por tanto, la pendiente de la recta resulta negativa): (14)")
59
Figura 4

60
Un ajuste por mínimos cuadrados, a una recta del tipo y = a+b x , nos permitirá decidir si la suposición realizada al admitir orden es correcta. Por otra parte, despejando [A] de la ecuación (14) se obtiene (15) donde podemos apreciar que la concentración de A, si la reacción es de primer orden y A es un reactivo, decrece exponencialmente con el tiempo (no olvidemos que si A es reactivo νA < 0 y que la constante de velocidad k siempre es positiva). Si la reacción química fuera elemental, al ser de primer orden tendría que ser monomolecular y entonces el coeficiente estequiométrico νA sería la unidad. Si la reacción no es elemental, puede ser de orden 1 y tener un coeficiente estequiométrico νA distinto de 1.
![Un ajuste por mínimos cuadrados, a una recta del tipo y = a+b x , nos permitirá decidir si la suposición realizada al admitir orden es correcta. Por otra parte, despejando [A] de la ecuación (14) se obtiene](http://slideplayer.es/slide/27139/1/images/60/Un+ajuste+por+m%C3%ADnimos+cuadrados%2C+a+una+recta+del+tipo+y+%3D+a%2Bb+x+%2C+nos+permitir%C3%A1+decidir+si+la+suposici%C3%B3n+realizada+al+admitir+orden+es+correcta.+Por+otra+parte%2C+despejando+%5BA%5D+de+la+ecuaci%C3%B3n+%2814%29+se+obtiene.jpg "(15) donde podemos apreciar que la concentración de A, si la reacción es de primer orden y A es un reactivo, decrece exponencialmente con el tiempo (no olvidemos que si A es reactivo νA < 0 y que la constante de velocidad k siempre es positiva). Si la reacción química fuera elemental, al ser de primer orden tendría que ser monomolecular y entonces el coeficiente estequiométrico νA sería la unidad. Si la reacción no es elemental, puede ser de orden 1 y tener un coeficiente estequiométrico νA distinto de 1.")
61
Un concepto relacionado con las ecuaciones de velocidad integradas es el tiempo de semirreacción, t1/2, el cual se define como el tiempo que debe transcurrir para que la concentración inicial de reactivo A se reduzca a la mitad; es decir, Llevando los datos anteriores a la ecuación (14) y teniendo en cuenta que A debe ser un reactivo (νA < 0 y por tanto νA = - | νA | ) tendremos de la expresión (16) se aprecia que el tiempo de semireacción, para una reacción que obedezca a una cinética de primer orden, es independiente de la concentración inicial de reactivo A. (16)
 y teniendo en cuenta que A debe ser un reactivo (νA < 0 y por tanto νA = - | νA | ) tendremos. de la expresión (16) se aprecia que el tiempo de semireacción, para una reacción que obedezca a una cinética de primer orden, es independiente de la concentración inicial de reactivo A. (16)")
62
Gráficamente:

63
2-Cinética de segundo orden
Las dos formas más comunes de ecuaciones cinéticas de segundo orden son donde A y B son dos reactivos diferentes (téngase en cuenta que si las concentraciones de A y B fueran iguales en todo instante, la forma de la segunda ecuación cinética sería la misma que la de la primera). Estudiemos ambos casos por separado.
. Estudiemos ambos casos por separado.")
64
2.1- En la reacción interviene un solo reactivo A o dos reactivos A y B con idénticas concentraciones en todo instante. Para la reacción νA→ Productos, o para la reacción νA+ νB →Productos, si se trata de una cinética de segundo orden, se tendrá la siguiente ecuación de velocidad: Separando variables e Integrando (17)
")
65
Una representación gráfica de 1/C frente al tiempo nos da una recta de pendiente positiva igual a k Figura 5

66
Para hallar t1/2 sustituyamos C por C0/2 y t por t1/2 en la expresión (17). Así tenemos
(18) Por tanto, para este tipo de reacciones de segundo orden, el tiempo de semirreacción t1/2 es inversamente proporcional a la concentración inicial del reactivo, a diferencia de las cinéticas de primer orden donde t1/2 era independiente de la concentración inicial de reactivo.
 Por tanto, para este tipo de reacciones de segundo orden, el tiempo de semirreacción t1/2 es inversamente proporcional a la concentración inicial del reactivo, a diferencia de las cinéticas de primer orden donde t1/2 era independiente de la concentración inicial de reactivo.")
67
2.2. En la reacción intervienen dos reactivos A y B con distintas concentraciones en el transcurso de la reacción. En este caso los coeficientes estequiométricos de A y B pueden ser iguales o distintos, pero si son iguales entonces necesariamente las concentraciones iniciales de A y B deben ser distintas. Si los coeficientes estequiométricos fueran distintos, podríamos partir de concentraciones iniciales iguales y, a pesar de ello, las concentraciones de A y B, en el transcurso de la reacción, serían distintas. t>0 o t=t a-x b-x

68
La ecuación de velocidad
expresada en función de x, resulta para el caso particular de que νA = νB= -1 (19)
")
69
3. Cinética de orden n. Consideremos únicamente una de las múltiples posibilidades que pueden darse. Supongamos que la reacción es de orden n y su ecuación cinética puede, por tanto, escribirse en la forma

70
Separando variables e integrando
(20) Obsérvese que la anterior ecuación (20) puede ser particularizada para cualquier valor de n excepto n = 1. Para una reacción de orden n, como las del tipo que estamos considerando, podemos obtener el tiempo de semirreacción sustituyendo en (20) la concentración C por C0/2 y el tiempo t por t1/2. Haciendo lo indicado y despejando t1/2 tendremos (21)
 Obsérvese que la anterior ecuación (20) puede ser particularizada para cualquier valor de n excepto n = 1. Para una reacción de orden n, como las del tipo que estamos considerando, podemos obtener el tiempo de semirreacción sustituyendo en (20) la concentración C por C0/2 y el tiempo t por t1/2. Haciendo lo indicado y despejando t1/2 tendremos. (21)")
71
Comparación de ambos métodos
El método de integración es probablemente el que más se usa en la interpretación de los datos cinéticos. Su principal inconveniente es que hay que “probar”: primero se supone cuál puede ser el orden y luego se comprueba si ese orden responde a los resultados experimentales. Una de las consecuencias de aplicar este método es que se tiende a laidea de que el orden debe ser un número entero. Si el orden es, por ejemplo 1.8, los resultados probablemente se ajustarán bien a una ecuación de segundo grado, quedando inadvertidas las desviaciones respecto a las características de este orden. Actualmente, está prácticamente superado este problema, dado que si se hace un programa simple, se puede determinar el valor de la pendiente (entera o decimal) con muy buena exactitud y en pocos minutos. El método diferencial es más correcto teóricamente. Su principal inconveniente es, a veces, que las pendientes no se pueden determinar con mucha precisión.
 con muy buena exactitud y en pocos minutos. El método diferencial es más correcto teóricamente. Su principal inconveniente es, a veces, que las pendientes no se pueden determinar con mucha precisión.")
72
Descomposiciones unimoleculares. Mecanismo de Lindemann.
Antes de 1922, la existencia de descomposiciones unimoleculares presentaba un serio problema de interpretación. La etapa unimolecular elemental consiste en el rompimiento de una molécula en fragmentos: AProductos Si esto ocurre, es porque el contenido energético de la molécula es demasiado grande. Demasiada energía se transforma, por alguna razón, en un grado particular de frecuencia vibratoria y por tanto esta vibración ocaciona o produce la disociación de la molécula en fragmentos. Las moléculas que tienen exceso de energía se descomponen.

73
Si la descomposición es contínua, otras moléculas deben ganar un exceso de energía. El problema se presenta cuando nos preguntamos cómo adquieren las moléculas esta cantidad extra de energía. Parecia que las moléculas no podían adquirir la energía necesaria mediante colisiones con otras moléculas, ya que la frecuencia de las colisiones depende del cuadrado de la concentración, esto haría que la reacción fuera de segundo orden, mientras se observa que era de primer orden. En 1922 Lindemann propuso un mecanismo según el cual las moléculas podían activarse por colisión y, a pesar de ello, la reacción podía ser de primer orden. La activación de las moléculas por colisión es:

74
Donde A es una molécula normal y A. es una molécula activada
Donde A es una molécula normal y A* es una molécula activada. La colisión entre dos moléculas normales, genera una molécula activada A* que tiene un exceso de energía en los varios grados vibratorios de libertad, la molécula remanente es deficiente en energía Una vez formada la molécula activada, puede seguir uno de los dos caminos siguientes: a- puede inactivarse por colisión, O b- puede transformarse en productos

75
La velocidad de formación de productos:
(22) En esta ecuación se presenta el problema de expresar la concentración de una especie activa en función de la concentración de especies normales. Una forma de resolver esto es aplicando la aproximación de régimen estacionario (o del estado estacionario). Se asume que la velocidad de formación de moléculas activadas es igual a la velocidad de desaparición, de forma que la velocidad final de cambio de concentración de A* es cero. Para justificar este argumento se puede decir que una vez comenzada la reacción, se alcanza un estado estacionario en el cual la concentración de las moléculas activas no cambian mucho de modo que El valor de la concentración de A* es pequeño y por lo tanto el cambio de concentración se asume cero sin cometer demasiado error. Así se tiene
 En esta ecuación se presenta el problema de expresar la concentración de una especie activa en función de la concentración de especies normales. Una forma de resolver esto es aplicando la aproximación de régimen estacionario (o del estado estacionario). Se asume que la velocidad de formación de moléculas activadas es igual a la velocidad de desaparición, de forma que la velocidad final de cambio de concentración de A* es cero. Para justificar este argumento se puede decir que una vez comenzada la reacción, se alcanza un estado estacionario en el cual la concentración de las moléculas activas no cambian mucho de modo que . El valor de la concentración de A* es pequeño y por lo tanto el cambio de concentración se asume cero sin cometer demasiado error. Así se tiene.")
76
Este valor de concentración, lleva a la ecuación (22) a la forma:
Hay dos situaciones límites importantes de la ecuación (23) Caso 1: Supongamos que la velocidad de descomposición, es sumamente alta, tan alta que tan pronto se forma la especie activa se descompone. (23)
 Caso 1: . Supongamos que la velocidad de descomposición, es sumamente alta, tan alta que tan pronto se forma la especie activa se descompone. (23)")
77
No hay tiempo para que se produzca una colisión de desactivación y la rapidez de desactivación es muy pequeña comparada con la descomposición. Entonces ó Por tanto, el denominador Y la ecuación (23) La velocidad de la reacción es igual a la rapidez con que se forman las moléculas activadas, ya que estas se descomponen inmediatamente. La cinética es de segundo orden. (24)
 La velocidad de la reacción es igual a la rapidez con que se forman las moléculas activadas, ya que estas se descomponen inmediatamente. La cinética es de segundo orden. (24)")
78
Caso Si despues de la activación hay una apreciable cantidad de tiempo antes de que la molécula se descomponga, hay oportunidad de que la molécula activa sufra colisiones que la desactiven Si el tiempo es considerable, entonces la rapidez de desactivación, , es mucho mayor que la de descomposición Esto significa que : Así, la ecuación (23) será: Y la ecuación de velocidad es de primer orden. El destino usual de la molécula activa al sufrir colisiones es la desactivación. Sólo una pequeña fracción de moléculas activas se descompone para dar productos. (25)
 será: Y la ecuación de velocidad es de primer orden. El destino usual de la molécula activa al sufrir colisiones es la desactivación. Sólo una pequeña fracción de moléculas activas se descompone para dar productos. (25)")
79
En una reacción en fase gaseosa, una presión alta aumenta el número de colisiones, de modo que es grande y la rapidez es de primer orden. El siministro de moléculas activas es adecuado y la rapidez a la cual se descomponen, limita la velocidad de la reacción. A presiones bajas disminuye el número de colisiones, es pequeño, y la rapidez es de segundo orden. La velocidad depende de la rapidez a la cual se producen moléculas activas por colisiones. El mecanismo de Lindemann se acepta como el mecanismo de activación de la molécula.

80
Aproximación del estado estacionario (steady state):
Establece que en condiciones estacionarias la concentración del intermediario es siempre mucho menor que la concentración de los reaccionantes, [X] alcanzará rápidamente un valor que permanecerá prácticamente constante durante todo el curso de la reacción. Por ejemplo para la reacción:

81
Para k2>>k1:

82
v = k [A] [B] Velocidad de reacción Factores que modifican k:
A y B reactivos k = constante de velocidad de reacción Factores que modifican k: Temperatura La temperatura y los catalizadores afectan reacciones en fase gaseosa y en solución El solvente y la fuerza iónica afectan reacciones en solución Catalizadores Solvente Fuerza iónica
![v = k [A] [B] Velocidad de reacción Factores que modifican k:](http://slideplayer.es/slide/27139/1/images/82/v+%3D+k+%5BA%5D%EF%81%A1+%5BB%5D+%EF%81%A2+Velocidad+de+reacci%C3%B3n+Factores+que+modifican+k%3A.jpg "A y B reactivos. k = constante de velocidad de reacción. Factores que modifican k: Temperatura. La temperatura y los catalizadores afectan reacciones en fase gaseosa y en solución. El solvente y la fuerza iónica afectan reacciones en solución. Catalizadores. Solvente. Fuerza iónica.")
83
Efecto de la temperatura sobre la velocidad de reacción
La ecuación de velocidad o su forma no varía con la temperatura pero sí la constante de velocidad, que aumenta al aumentar la temperatura. Empíricamente se encontró que la “k” está relacionada con la temperatura en la forma donde A y B son constantes y aplicando logaritmo y pasando a log10 (26) (27)
 (27)")
84
que gráficamente daría:
Log k 1/T

85
En 1887 Van’T Hoff consideró que la constante k podría interpretarse su dependencia con la temperatura en la misma forma que la constante de equilibrio: K=f (T) definida por él En el año 1889 (dos años más tarde) esta idea fue desarrollada por ARRHENIUS y aplicada con éxito a muchas reacciones y para lo cual teniendo en cuenta lo considerado por Van’t Hoff. Actualmente la influencia de la temperatura en la velocidad de las reacciones químicas se interpreta generalmente en términos de la expresión conocida como ecuación de Arrhenius, según la cual la constante de velocidad es el producto de un factor pre-exponencial (A) y un factor exponencial
 esta idea fue desarrollada por ARRHENIUS y aplicada con éxito a muchas reacciones y para lo cual teniendo en cuenta lo considerado por Van’t Hoff. Actualmente la influencia de la temperatura en la velocidad de las reacciones químicas se interpreta generalmente en términos de la expresión conocida como ecuación de Arrhenius, según la cual la constante de velocidad es el producto de un factor pre-exponencial (A) y un factor exponencial.")
86
Efecto de la temperatura sobre la velocidad de reacción
Ecuación de Arrhenius Constante de velocidad factor pre-exponencial o factor de frecuencia factor exponencial o factor de Boltzmann k = A . e –Ea/RT (28) El factor exponencial depende de la temperatura, la constante de los gases (R= J mol-1 k-1) y la energía de activación (Ea). Las unidades de A se corresponden a las de k. Ea = energía de activación R = constante de los gases T = temperatura absoluta
 El factor exponencial depende de la temperatura, la constante de los gases (R= J mol-1 k-1) y la energía de activación (Ea). Las unidades de A se corresponden a las de k. Ea = energía de activación. R = constante de los gases. T = temperatura absoluta.")
87
Energía de activación (Ea) A + B Productos
Es la energía cinética mínima que deben alcanzar los reactivos para que la reacción sea efectiva y evolucione a los productos A + B P Ea-1 Ea1 U A + B Productos Ea1 = Ea de la reacción directa Ea-1 = Ea de la reacción inversa U = Ea1 – Ea-1 (reacciones elementales) Energía Trayectoria de la reacción
 Energía. Trayectoria de la reacción.")
88
Existen además otras expresiones empíricas para el estudio de la dependencia de k con la temperatura:

89
Para hallar la ecuación de Arrhenius:
Si consideramos una reacción reversible del tipo: De acuerdo a la ley de Goldberg y Waage: en el equilibrio aplicando logaritmos a K y derivando con respecto a T y teniendo en cuenta la ecuación de Van´T Hoff (29)
")
90
la ecuación (29) puede desdoblarse en dos ecuaciones y teniendo en cuenta que los subíndices (-1), corresponden a los términos de la reacción inversa: (30) integrando las ecuaciones (30) y eliminando logaritmos se tiene (31)
 integrando las ecuaciones (30) y eliminando logaritmos se tiene. (31)")
91
El factor de frecuencia tiene las mismas unidades que k, por cuanto es adimensional y dividiendo las ecuaciones (31) , darán la constante de equilibrio K que concuerda con la ecuación de VAN’T HOFF: k= f(T) en el equilibrio Esto no significa que H sea igual a Ea, sino que ambos términos representan diferencias de energía, en el primer caso (H) calor de reacción y en el segundo (Ea) la de las energías de las moléculas reactantes y productos de la reacción, cuyo concepto lo daremos más adelante. “ESTA LEY DE ARRHENIUS ES DE VALIDEZ UNIVERSAL”, para las reacciones elementales, una desviación de la ley es, de hecho, señal que la reacción no es sencilla.
 en el equilibrio. Esto no significa que H sea igual a Ea, sino que ambos términos representan diferencias de energía, en el primer caso (H) calor de reacción y en el segundo (Ea) la de las energías de las moléculas reactantes y productos de la reacción, cuyo concepto lo daremos más adelante. ESTA LEY DE ARRHENIUS ES DE VALIDEZ UNIVERSAL , para las reacciones elementales, una desviación de la ley es, de hecho, señal que la reacción no es sencilla.")
92
Efecto de la temperatura sobre la velocidad de reacción
Ecuación de Arrhenius (32) k = A . e –Ea/RT Isocora de Van’t Hoff K ln = T d 2 RT DH es análoga a: DU K ln = T d 2 RT Para fases condensadas H = U
 k = A . e –Ea/RT. Isocora de Van’t Hoff. K. ln. = T. d. 2. RT. DH. es análoga a: DU. K. ln. = T. d. 2. RT. Para fases condensadas H = U.")
93
Uso de la ecuación de Arrhenius
Se utiliza para predecir como cambia la velocidad de reacción a distintas temperaturas (Ej: descomposición de fármacos) Ecuación empírica, no exacta k = A . e –Ea/RT ln k m = -Ea/R 1/T
 Ecuación empírica, no exacta. k = A . e –Ea/RT. ln k. m = -Ea/R. 1/T.")
94
Limitaciones de la ecuación de Arrhenius
Buena aproximación de la dependencia de k con la T (si el rango de T es chico) Una desviación de la linealidad indicaría la presencia de una reacción compleja. Pero, una representación lineal, no indica que la reacción sea una reacción elemental Representación no lineal indica que Ea y A no son independientes de la T.
 Una desviación de la linealidad indicaría la presencia de una reacción compleja. Pero, una representación lineal, no indica que la reacción sea una reacción elemental. Representación no lineal indica que Ea y A no son independientes de la T.")
95
k = P. 2AB . (8 kB T/ )1/2 . e-Ea/RT
Modelos para calcular la constante de velocidad (k) de reacciones químicas elementales v = k [A] [B] k = A . e –Ea/RT Teorías sobre los mecanismos de reacción de los procesos elementales Teoría de Arrhenius Teoría de las colisiones k = P. 2AB . (8 kB T/ )1/2 . e-Ea/RT A Teoría del complejo activado
 de reacciones químicas elementales. v = k [A] [B] k = A . e –Ea/RT. Teorías sobre los mecanismos de reacción de los procesos elementales. Teoría de Arrhenius. Teoría de las colisiones. k = P. 2AB . (8 kB T/ )1/2 . e-Ea/RT. A. Teoría del complejo activado.")
96
TEORÍA DE ARRHENIUS (1889) Desarrolló el primer estudio teórico, que es una descripción cualitativa, para explicar el comportamiento molecular que corresponde a la forma obtenida experimentalmente para la constante de velocidad. Se va a utilizar la reacción reversible, y muy sencilla, entre el vapor de yodo y el hidrógeno, para formar yoduro de hidrógeno, con el fin de exponer las ideas de Arrhenius. Esta reacción H2 + I2 2HI es de segundo orden, tanto en sentido directo como el inverso, y aparentemente se desarrolla en un solo paso por un proceso de cuatro centros, de modo que el camino de la reacción se puede representar así:

97
Para sistemas más complicados, aún cuando su mecanismo ya esté establecido, no es fácil ver cómo los electrones y átomos se desplazan mientras la reacción se desarrolla. Aunque Arrhenius, pese a todo, reconoció que cualquier proceso de reacción se puede admitir que procede al menos formando primero una especie de alta energía, que en la actualidad se llama complejo activado, y en segundo lugar produciéndose la disociación del complejo en los productos finales.

98
Si se supone que el complejo activado requiere para formarse una energía Ea superior a la que poseen los reactivos, el número de moléculas complejos activos, comparado con el número de moléculas reactivas, puede estimarse con la ley de distribución de Boltzmann, en la forma: (33) dividiendo por el volumen, el numerador y denominador la (33) quedará (34)
 dividiendo por el volumen, el numerador y denominador la (33) quedará. (34)")
99
Arrhenius, postuló que la velocidad de la reacción es proporcional al número o concentración de los complejos activos, denominando con A al factor de proporcionalidad, expresó la siguiente relación: (35) reemplazando la concentración de moléculas activadas de acuerdo a lo expresado anteriormente y que según la (35), la ecuación de velocidad quedará (36) donde se observa que conduce al mismo resultado encontrado en las ecuaciones de velocidad y de esta forma se llegaría a la relación:
 reemplazando la concentración de moléculas activadas de acuerdo a lo expresado anteriormente y que según la (35), la ecuación de velocidad quedará. (36) donde se observa que conduce al mismo resultado encontrado en las ecuaciones de velocidad y de esta forma se llegaría a la relación:")
100
La teoría de Arrhenius solo describe cualitativamente una reacción, pero no puede demostrar como A depende de las propiedades de las moléculas en reacción y no permite determinar en forma teórica, el valor de Ea. La hipótesis de un complejo activado puede representarse esquemáticamente en un gráfico que muestra la energía del sistema, medida en ordenadas, en relación con la coordenada de reacción, que figura como abscisa. La coordenada de reacción, no es precisamente una distancia internuclear cualquiera, sino que más bien depende de todas las distancias internucleares que experimentan variación a medida que los reactivos se convierten en productos

101
Perfil de una reacción Reac. exotérmica Reac. endotérmica Entalpía
Complejo activado Complejo activado Energía de activación productos reactivos reactivos productos Entalpía de reacción (H) Coordenada de reacción Reac. exotérmica Reac. endotérmica
 Coordenada de reacción. Reac. exotérmica. Reac. endotérmica.")
102
TEORÍA DE LAS COLISIONES-Modelo colisional
Las moléculas (reactivos) deben colisionar para reaccionar La velocidad de reacción es proporcional al número de colisiones por unidad de tiempo y unidad de volumen y a la fracción de choques. Las moléculas reaccionan si: - orientación molecular favorable (requisito estérico) - E cinética colisión Ea (requisito energético) La Ecinética de las moléculas se describe por la distribución de energías de Boltzmann la colisión puede requerir una orientación especial para ser reactiva
 deben colisionar para reaccionar. La velocidad de reacción es proporcional al número de colisiones por unidad de tiempo y unidad de volumen y a la fracción de choques. Las moléculas reaccionan si: - orientación molecular favorable (requisito estérico) - E cinética colisión Ea (requisito energético) La Ecinética de las moléculas se describe por la distribución de energías de Boltzmann. la colisión puede requerir una orientación especial para ser reactiva.")
103
Según esta teoría, para que ocurra una reacción química es necesario que existan choques entre las moléculas de reactantes que den origen a productos. Estas colisiones deben cumplir las siguientes condiciones: Las moléculas de reactantes deben poseer la energía suficiente para que pueda ocurrir el rompimiento de enlaces, reordenamiento de los átomos y luego la formación de los productos. Si no se dispone de la energía suficiente, las moléculas rebotan sin formar los productos. Los choques entre las moléculas deben efectuarse con la debida orientación en los reactantes Si el choque entre las moléculas cumple con estas condiciones, se dice que las colisiones son efectivas y ocurre la reacción entre los reactantes; entonces se forman productos. Cabe destacar que no todas las colisiones entre reactantes son efectivas, por lo tanto no todas originan productos. Sin embargo, mientras más colisiones existan entre reactantes, mayor es la probabilidad de que sean efectivas

104
Requisito estérico Cl + NOCl Cl2 + NO
Orientación molecular adecuada Orientación molecular inadecuada

105
Requisito energético H2 + Cl2 2 HCl Ec Ea
Solo una pequeña fracción de colisiones son efectivas Ec Ea

106
Si se considera la reacción: A + B productos Es necesario calcular el número de choques que se producen por unidad de tiempo y por centímetros cúbicos, de las contenidas en el reactor y como este cálculo esta realizado para los gases, “La Teoría de las Colisiones” está restringida, por tanto, a la interpretación de las reacciones en fase gaseosa. Recordando que de acuerdo a la teoría cinético molecular de los gases el número de choques por unidad de tiempo y de volumen es: (37)
")
108
Si para simplificar se supone que las moléculas de A y B tienen pesos moleculares y diámetros muy parecidos la ecuación (37) quedará: (38) y como además solo interesan los choques entre las moléculas de A con B, debemos restar del número total de choques, los choques totales de A con A, y de B con B y si denominamos con ZA, ZB y ZAB los respectivos choques de las moléculas de A, B y AB, entre sí, lo que interesa son los choques de A y B que pueden adquirir la energía suficiente para generar productos, por tanto: (39) (40)
 y como además solo interesan los choques entre las moléculas de A con B, debemos restar del número total de choques, los choques totales de A con A, y de B con B y si denominamos con ZA, ZB y ZAB los respectivos choques de las moléculas de A, B y AB, entre sí, lo que interesa son los choques de A y B que pueden adquirir la energía suficiente para generar productos, por tanto: (39) (40)")
109
resolviendo el cuadrado del binomio y eliminando los términos donde aparecen por ser iguales y de signo contrario la ecuación (40) quedará: (41) Para diámetros distintos y M distintos debemos tomar los promedios de los diámetros moleculares: y de los pesos moleculares mediante la masa reducida y por tanto la ecuación (41) quedará expresada da la siguiente forma: (42)
 Para diámetros distintos y M distintos debemos tomar los promedios de los diámetros moleculares: y de los pesos moleculares mediante la masa reducida y por tanto la ecuación (41) quedará expresada da la siguiente forma: (42)")
110
introduciendo el denominador 4 y dentro de la raíz:
(43) Para no contar dos veces una colisión entre la molécula A y B se elimina el factor raiz de 2 (44)
 Para no contar dos veces una colisión entre la molécula A y B se elimina el factor raiz de 2. (44)")
111
Según Lewis, como se dijo al principio, la velocidad es proporcional al número de choques efectivos entre A y B y a la fracción de choques entre éstas que adquieren por choques suficiente energía para conseguir la reorganización de átomos y electrones, que dará lugar a moléculas producto de reacción, por lo tanto: (45) Habiendo determinado ZAB, ahora es necesario calcular la fracción de choques y para una mayor sencillez se esquematiza uno de los choques efectivos (choque con reacción) suponiendo que la energía de traslación de un grado de libertad de cada molécula, se convierte en el momento del choque en otra forma de energía del sistema formado por las dos moléculas, que están colisionando y que la energía adquirida por los dos grados de libertad (de las dos moléculas) es suficiente para superar la barrera de energía que impide la reacción
 Habiendo determinado ZAB, ahora es necesario calcular la fracción de choques y para una mayor sencillez se esquematiza uno de los choques efectivos (choque con reacción) suponiendo que la energía de traslación de un grado de libertad de cada molécula, se convierte en el momento del choque en otra forma de energía del sistema formado por las dos moléculas, que están colisionando y que la energía adquirida por los dos grados de libertad (de las dos moléculas) es suficiente para superar la barrera de energía que impide la reacción.")
112
Ahora se pueden realizar los cálculos en base a una distribución bidimensional de las velocidades moleculares ya que participan en cada choque efectivo dos moléculas de especie química diferente y que vienen dada por una ley de distribución equivalente a la de las velocidades mono y tridimensional. Esta distribución es: (46)
")
113
Distribución de energías de Boltzmann
A mayor temperatura, mayor número de colisiones con E Ea

114
como Nav.m=M y Nav.k=R, queda:
Esta ecuación se puede escribir en función de las energías por mol, esto es, de la energía de un número de moléculas N’, igual al de Avogadro (Nav), y para ello se multiplica y divide por el Nav numerador y denominador del factor del exponencial y del exponente como Nav.m=M y Nav.k=R, queda: (47) Y siendo la Energía cinética por mol: éstas expresiones conducen a que la (47) quede expresada en la siguiente forma
, y para ello se multiplica y divide por el Nav numerador y denominador del factor del exponencial y del exponente. como Nav.m=M y Nav.k=R, queda: (47) Y siendo la Energía cinética por mol: éstas expresiones conducen a que la (47) quede expresada en la siguiente forma.")
115
Si se integra (48) reemplazando las ecuaciones (44) y (48) en la (45) se tiene que
 reemplazando las ecuaciones (44) y (48) en la (45) se tiene que")
116
Al comparar esta ecuación con la que se obtuvo experimentalmente para la velocidad de reacción, si ésta última se expresa en función de las concentraciones molares cA y cB en lugar de una función de moléculas por centímetro cúbico, es decir Se obtiene: (49)
")
117
Este resultado corresponde a la ecuación de velocidad de una reacción de segundo orden entre A y B, si la constante de velocidad se supone que es Este valor lo podemos comparar con la constante de velocidad de la ecuación de Arrhenius, el factor preexponencial, A, equivale y puede evaluarse con la relación: El resultado más satisfactorio de la teoría de las colisiones, es poder calcular el valor de A y a su vez comprobar con el obtenido experimentalmente.

119
Modelo del complejo activado
Estado de transición Complejo activado Arreglo particular de átomos que corresponde al máximo valor de energía (1 configuración) Toda configuración que se encuentra en la región cercana al máximo de la barrera energética Estado de transición Complejo activado Energía A + B P Trayectoria de la reacción
 Toda configuración que se encuentra en la región cercana al máximo de la barrera energética. Estado de transición. Complejo activado. Energía. A + B. P. Trayectoria de la reacción.")
120
Modelo del complejo activado (2)
A B P K# k# A B AB# P La reacción química es el resultado del paso del complejo activado sobre la barrera energética La velocidad de reacción es proporcional al número de complejos activados que pasan sobre la barrera energética por unidad de tiempo El complejo activado (AB#) se encuentra en equilibrio con los reactivos La descomposición del [AB#] - sigue una cinética de primer orden (k#) - limita y determina la velocidad de reacción
 se encuentra en equilibrio con los reactivos. La descomposición del [AB#] - sigue una cinética de primer orden (k#) - limita y determina la velocidad de reacción.")
121
Modelo del complejo activado
Reacción que ocurre a través de un mecanismo SN2

122
La velocidad de reacción depende de dos factores: la concentración de estados de transición y la velocidad con que éstos se rompen para formar los productos de reacción. La concentración de complejos activados, al menos formalmente, puede establecerse en función de la ecuación de equilibrio: (50) La velocidad con que el complejo se rompe se puede estimar, suponiendo que se escindirá en las moléculas de los productos, cuando una adecuada vibración, con una amplitud suficientemente grande, ayuda a distender el complejo hasta su ruptura. La frecuencia de esta vibración, por tanto será en cierto modo similar a la velocidad con que el complejo se disocia.
 La velocidad con que el complejo se rompe se puede estimar, suponiendo que se escindirá en las moléculas de los productos, cuando una adecuada vibración, con una amplitud suficientemente grande, ayuda a distender el complejo hasta su ruptura. La frecuencia de esta vibración, por tanto será en cierto modo similar a la velocidad con que el complejo se disocia.")
123
El complejo activo es una sustancia inestable y se mantiene unido por enlaces muy lábiles. Por consiguiente, las vibraciones de estas moléculas han de tener frecuencias muy bajas y la energía media por grado de libertad de vibración será aproximadamente de la cuantía de la energía kT. Por tanto, al aplicar la relación de Planck: al modo de vibración que produce la ruptura del complejo, su frecuencia de vibración se estima en: Se debe indicar que con este razonamiento se alcanza un valor de: (51)
")
124
Y que este valor, es para enlaces químicos lábiles.
Por tanto, la velocidad de reacción en la teoría del estado de transición es: Al comparar con las ecuaciones empíricas de velocidad para las reacciones de segundo orden, se demuestra que la constante de velocidad k2 se ha de identificar con (52) (53)
 (53)")
125
Al sustituir esta relación en la ecuación (54) se obtiene (55)
Este resultado tiene significado cuando la constante de equilibrio se interpreta termodinámicamente. Por este camino, se introducen los conceptos de energía libre de activación, entropía de activación y entalpía de activación La constante de equilibrio se relaciona con la energía libre de formación del complejo activo, de la siguiente forma: (54) Para la reacción a una temperatura determinada, la energía libre de activación puede considerarse resultado de una contribución de entalpía y otra de entropía, de acuerdo con: Al sustituir esta relación en la ecuación (54) se obtiene (55) (56)
 Para la reacción a una temperatura determinada, la energía libre de activación puede considerarse resultado de una contribución de entalpía y otra de entropía, de acuerdo con: Al sustituir esta relación en la ecuación (54) se obtiene. (55) (56)")
126
Con este valor de K se deduce para la constante de velocidad:
(57) la cual, recordando que la variación que determina el factor T es pequeña comparada con el valor del factor exponencial, concuerda con la ecuación de Arrhenius. El factor exponencial contiene ahora una entalpía en lugar de la energía de activación. En los sistemas líquidos, como se estudió, la diferencia debe ser del todo despreciable y en los sistemas gaseosos, aunque resulta pequeña, es necesario calcularla a partir de RTn. De nuevo es la interpretación teórica del factor preexponencial A lo que presenta más destacado interés.
 la cual, recordando que la variación que determina el factor T es pequeña comparada con el valor del factor exponencial, concuerda con la ecuación de Arrhenius. El factor exponencial contiene ahora una entalpía en lugar de la energía de activación. En los sistemas líquidos, como se estudió, la diferencia debe ser del todo despreciable y en los sistemas gaseosos, aunque resulta pequeña, es necesario calcularla a partir de RTn. De nuevo es la interpretación teórica del factor preexponencial A lo que presenta más destacado interés.")
127
ENTROPÍA DE ACTIVACIÓN
La teoría del estado de transición interpreta el factor preexponencial A de la ecuación de Arrhenius en la forma: y por tanto, requiere una evaluación de la entropía de activación, antes de realizar el cálculo teórico de la magnitud A. Dado que (S0) es la variación de entropía al transformarse los reactivos en el complejo activado, y como se conoce poco de las propiedades de este complejo, la teoría del estado activado tiende a evitar predicciones acerca del valor de cualquier propiedad determinada del mismo. (58)
 es la variación de entropía al transformarse los reactivos en el complejo activado, y como se conoce poco de las propiedades de este complejo, la teoría del estado activado tiende a evitar predicciones acerca del valor de cualquier propiedad determinada del mismo. (58)")
128
En las reacciones en fase gaseosa, como es la formación del ioduro de hidrógeno a partir de yodo e hidrógeno, se reconoce que, cuando las moléculas reactivos se unen una a otra para formar el estado de transición, el número total de grados de libertad de traslación disminuye de 6 a 3 y el de rotación de 4 a 3 (o de 6 a 3 sí las dos moléculas reactivo no son lineales). Los grados de libertad que se pierden se transforman en 4 (ó 6) grados de libertad de vibración del complejo activado. No es cuestión que se pueda estudiar de qué tipo son los débiles enlaces en el estado de transición, pero es indudable que ha de haber pérdida de grados de libertad de traslación y rotación y aparición de nuevas vibraciones.
 grados de libertad de vibración del complejo activado. No es cuestión que se pueda estudiar de qué tipo son los débiles enlaces en el estado de transición, pero es indudable que ha de haber pérdida de grados de libertad de traslación y rotación y aparición de nuevas vibraciones..")
129
Los niveles de energía del complejo activo, estarán espaciados más ampliamente y la entropía del complejo debe ser menor que la de las moléculas reactivo. Los factores preexponenciales experimentales de unas cuantas reacciones similares a la reacción yodo-hidrógeno conducen siempre, mediante la ecuación (58), a, valores negativos de (S0), de acuerdo con las consideraciones precedentes. Sin embargo, si la rigidez impuesta por el complejo activo es pequeña, el cambio de entropía correspondiente debe ser pequeño
, a, valores negativos de (S0), de acuerdo con las consideraciones precedentes. Sin embargo, si la rigidez impuesta por el complejo activo es pequeña, el cambio de entropía correspondiente debe ser pequeño.")
130
Por esta razón, como se ve en la tabla 1, las diferencias entre los factores preexponenciales están realmente justificadas, aunque no se pueden calcular por las características del complejo activado o estado de transición .

131
Tabla 1: Constantes de velocidad de unas cuantas reacciones de segundo orden en fase gaseosa
Reacción Constante de velocidad(L/mol.seg) H2 + I2 2HI 2,0x109 2HI H2 + I2 3,3x109 2NO2 2NO + O2 2,6x108 2NOCl 2NO + Cl2 NO + Cl2 NOCl + Cl 1,0x108 CH3I + HI CH4 + I2 5,2x1010
 H2 + I2 2HI. 2,0x109. 2HI H2 + I2. 3,3x109. 2NO2 2NO + O2. 2,6x108. 2NOCl 2NO + Cl2. NO + Cl2 NOCl + Cl. 1,0x108. CH3I + HI CH4 + I2. 5,2x1010.")
Presentaciones similares