Descargar la presentación
La descarga está en progreso. Por favor, espere

1
Dr. Denis Padgett Moncada
Ética e investigación: Principios, procesos y papel de los comités de ética Dr. Denis Padgett Moncada Comisión de Ética CMH Y Coordinador del comité de Bioética de la UIC FCM

2
Registros Históricos Vacuna experimental de cólera termino en desastre por Plaga Iatrogénica en el presidio Manila Bilibid Filipinas 1933 Estudio de Sífilis de Tuskegee od/tuskegee/time.htm

3
1947 El Código de Nuremberg Dr. Josepf Mengele Ángel de la Muerte

4
1964 Declaración de Helsinki
La Declaración de Helsinki La Declaración de Helsinki fue publicada por primera vez por la Asociación Médica Mundial en Se cita comúnmente como una referencia en la mayoría de las normas nacionales o internacionales y es considerada por muchos como la primera norma internacional para la investigación biomédica. En el centro de la declaración figura la afirmación de que "el bienestar de los seres humanos debe tener siempre primacía sobre los intereses de la ciencia y de la sociedad". También se presta particular atención a la importancia del consentimiento informado por escrito. La Declaración de Helsinki ha sido modificada cinco veces, últimamente en el año 2001 para incluir temas de relevancia particular para el tipo de investigación que se está llevando a cabo actualmente, como el uso de los controles de placebo. Propone que todo nuevo método se someta a prueba contra los mejores métodos actuales comprobados profilácticos, diagnósticos o terapéuticos. La declaración modificada también afirma que la "investigación médica sólo se justifica si existen posibilidades razonables de que la población, sobre la que la investigación se realiza, podrá beneficiarse de los resultados de la investigación".

5
1976 El Reporte Belmont Codigoetica/codigosetica2.htm In November 1906, Richard Pearson Strong, then head of the Philippine Biological Laboratory, inoculated 24 men--inmates of Manila's Bilibid Prison--with a cholera vaccine that somehow had been contaminated with plague organisms; 13 men died. The governor-general of the Philippines appointed a general committee to investigate the affair, and the U.S. Senate demanded information about the episode. Although the Senate, the secretary of war, and even the president were kept informed of developments, no mainland investigations ensued. The general committee concluded that Strong was negligent for not having locks on his incubators and for leaving a visiting physician alone in the laboratory, where he might have mixed up the cholera and plague cultures on the fateful day. The committee's charge was referred to the attorney general, who found Strong innocent of criminal negligence, whereupon the governor-general exonerated Strong. Strong was despondent over Bilibid but recovered and developed a noteworthy career in American tropical medicine. In retrospect, the disaster at Bilibid presents an epitome of the problems surrounding the use of prisoner-subjects without authorization and without their voluntary consent.

6
Del Informe Belmont al Código de Reglamentos Federales
En 1972, el público estadounidense se enteró del estudio de Tuskegee sobre la Sífilis, en el cual se les negó tratamiento para la sífilis a 399 aparceros afroamericanos pobres del condado de Macon, en Alabama, quienes fueron engañados por los médicos del Servicio de Salud Pública de Estados Unidos desde 1932 hasta Como parte del estudio, diseñado para documentar la historia natural de la enfermedad, a estos hombres se les dijo que estaban recibiendo tratamiento por "sangre mala". En realidad, los funcionarios gubernamentales hicieron todo lo posible para asegurarse de que no recibieran ninguna terapia de ninguna fuente. El 26 de julio de 1972, el New York Times describió el estudio como "el experimento no terapéutico en seres humanos más largo en la historia clínica". La divulgación de este estudio por la prensa fue un gran escándalo en Estados Unidos. Para tratar este problema, en 1974 el gobierno de Estados Unidos estableció la Comisión Nacional para la Protección de Sujetos Humanos. En 1978, la comisión presentó su informe, el Informe Belmont: Principios éticos y pautas para la protección de sujetos humanos de la investigación. El informe abogó a favor del respeto por las personas, la beneficencia y la justicia como los principios fundamentales para la realización ética de investigaciones con participantes humanos. Los reglamentos federales actuales para la realización de investigaciones en Estados Unidos se basan en los principios éticos del Informe Belmont. El Código de los Reglamentos Federales, comúnmente conocido como 45 CFR 46 o Regla Común, expone las políticas básicas del gobierno de Estados Unidos para la protección de los seres humanos en la investigación. Incluye, entre otros, reglamentos detallados para los consejos de revisión institucional o los comités de la ética de la investigación y el consentimiento informado.

7
Normas del Consejo de Organizaciones Internacionales de las Ciencias Médicas
El Consejo de Organizaciones Internacionales de las Ciencias Médicas (CIOMS, por sus siglas en inglés), en colaboración con la Organización Mundial de la Salud, ha sido activo en la ética de la investigación por muchos años. El CIOMS emitió la primera publicación de sus Normas en 1993 y las modificó en Su finalidad fue: "preparar normas para indicar cómo los principios éticos que deben guiar la realización de la investigación biomédica en seres humanos, según se estipula en la Declaración de Helsinki, podrían aplicarse eficazmente, en particular en los países en desarrollo, dadas sus circunstancias socioeconómicas, sus leyes y sus reglamentos, así como los arreglos ejecutivos y administrativos". Las Normas del CIOMS reconocen el reto de aplicar los principios éticos universales en un mundo con recursos contrastantes. Las Normas consisten en 21 normas específicas, cada una seguida de observaciones interpretativas. Entre las normas de particular interés para los representantes comunitarios figuran: Comités de revisión ética Obtención de consentimiento informado: información esencial para futuros sujetos de investigación Beneficios para los participantes y sus comunidades Prestación de servicios de salud Distribución de las cargas y beneficios Las Normas del CIOMS son muy influyentes y se han difundido ampliamente. Se usan con frecuencia como una obra de consulta para formular las normas nacionales o locales.
, en colaboración con la Organización Mundial de la Salud, ha sido activo en la ética de la investigación por muchos años. El CIOMS emitió la primera publicación de sus Normas en 1993 y las modificó en Su finalidad fue: preparar normas para indicar cómo los principios éticos que deben guiar la realización de la investigación biomédica en seres humanos, según se estipula en la Declaración de Helsinki, podrían aplicarse eficazmente, en particular en los países en desarrollo, dadas sus circunstancias socioeconómicas, sus leyes y sus reglamentos, así como los arreglos ejecutivos y administrativos . Las Normas del CIOMS reconocen el reto de aplicar los principios éticos universales en un mundo con recursos contrastantes. Las Normas consisten en 21 normas específicas, cada una seguida de observaciones interpretativas. Entre las normas de particular interés para los representantes comunitarios figuran: Comités de revisión ética. Obtención de consentimiento informado: información esencial para futuros sujetos de investigación. Beneficios para los participantes y sus comunidades. Prestación de servicios de salud. Distribución de las cargas y beneficios. Las Normas del CIOMS son muy influyentes y se han difundido ampliamente. Se usan con frecuencia como una obra de consulta para formular las normas nacionales o locales.")
9
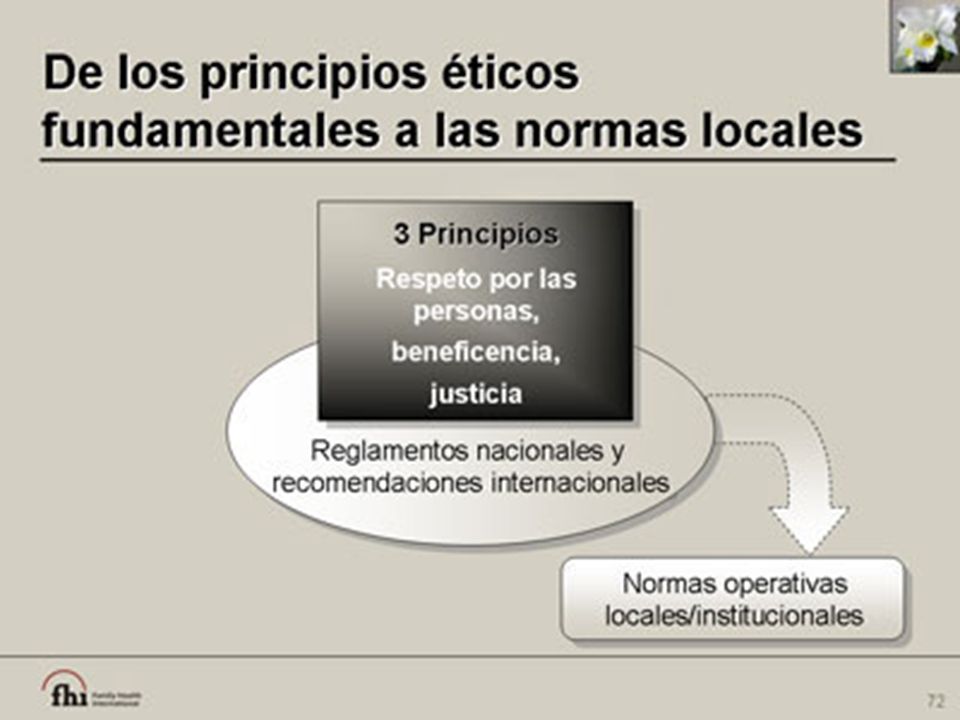
Principios Éticos Básicos
El Código de Nuremberg La Declaración de Helsinki, 1964 El Informe Belmont 1979. Las Directrices Éticas Internacionales para la Investigación Biomédica Relacionada con Seres Humanos La directriz de Buena Práctica Clínica de la OMS (1975) La Directriz de Buena Práctica Clínica de la Conferencia Internacional sobre Armonización 1996
 La Directriz de Buena Práctica Clínica de la Conferencia Internacional sobre Armonización")
11
Principios Éticos Básicos
Respeto por las Personas Respeto de la autonomía individual Es única y libre Derecho a decidir Protección a los individuos con autonomía reducida Derecho al Consentimiento Informado Privacidad y confidencialidad

13
Principios Éticos Básicos
Beneficencia Maximizar los beneficios y reducir los daños Análisis de riesgo / beneficio Mérito científicos Bienestar físico, psicológico y social del participante Retener las perspectivas de la comunidad

14
Justicia La justicia requiere la distribución justa y equitativa de los beneficios y riesgos de la participación en un estudio de investigación. El reclutamiento y la selección de los participantes deben hacerse de una manera justa y equitativa. La justicia prohíbe la exposición de un grupo de personas a los riesgos de la investigación exclusivamente para el beneficio de otro grupo. Los representantes comunitarios tienen la responsabilidad de garantizar que la participación de la comunidad en un estudio de investigación esté justificada. Los representantes comunitarios deben ser conscientes de la necesidad de proteger a los participantes debidamente durante la investigación. Deben prestar atención especial a los beneficios que los participantes o sus comunidades recibirán como resultado de su participación en la investigación e informar al equipo de investigación para que los incentivos ofrecidos por la investigación no influyan en la decisión de participar. El principio de la justicia establece protección especial para las personas vulnerables. La justicia no permite usar grupos vulnerables, tales como las personas de bajos recursos, como participantes en una investigación para el beneficio exclusivo de grupos más privilegiados

15
REQUISITOS DE LOS ESTUDIOS DE INVESTIGACIÓN
1.- Contar con los principios científicos generalmente aceptados. 2.- Contar con un protocolo de investigación que debe ser aprobado por un Comité de Ética. 3.- La investigación en seres humanos debe ser realizada sólo por personas científicamente calificadas, ó bajo la supervisión de un profesional competente. 4.- Los beneficios de la investigación para los individuos participantes deberán sobrepasar los riesgos involucrados en la participación.

16
REQUISITOS DE LOS ESTUDIOS DE INVESTIGACIÓN
5.-Deberá garantizar respeto a la integridad del ser humano sujeto de la investigación. 6.-Los resultados deberán darse a conocer con exactitud y apego a los hallazgos del proyecto. 7.-Obtener consentimiento del individuo que participará en la investigación, o de su representante legal en caso de no poder tomar la decisión por si mismo por ser menor de edad o tener alteraciones de la conciencia.

17
Elementos que constituyen un Estudio

19
Colaboradores en la investigación comunitaria: Un modelo
Se requiere la participación de muchos colaboradores para determinar qué tipo de investigación se debe llevar a cabo en una comunidad, así como para elaborarla, ejecutarla y difundir sus resultados. Este currículo se centra en una relación ideal entre tres de estos colaboradores. La comunidad donde se llevará a cabo la investigación, representada por los participantes de la investigación y otros miembros de la comunidad. El personal de investigación, contando al investigador del estudio y a otras personas con una función directa en la investigación. Se incluye también al patrocinador de la investigación, aunque esta persona u organización no siempre está situada físicamente en el lugar donde se está realizando la investigación. Por lo general, el patrocinador proporciona los fondos y recursos necesarios para realizar la investigación. (En la sección II se expone más información sobre el personal de investigación y los patrocinadores.) El comité de ética, que revisa y aprueba los procesos de investigación para garantizar que los participantes no sufran ningún daño en el estudio de investigación. (En la sección III se expone más información sobre los comités de ética.) Estos tres colaboradores deben trabajar de manera conjunta para garantizar que los estudios de investigación se lleven a cabo tomando en cuenta los mejores intereses de la comunidad. Todos los colaboradores tienen responsabilidades específicas entre sí y en cuanto al proceso de investigación. Es más probable que la investigación se lleve a cabo con éxito si todos los colaboradores cuentan con la información y capacitación necesarias para cumplir con sus responsabilidades.
 El comité de ética, que revisa y aprueba los procesos de investigación para garantizar que los participantes no sufran ningún daño en el estudio de investigación. (En la sección III se expone más información sobre los comités de ética.) Estos tres colaboradores deben trabajar de manera conjunta para garantizar que los estudios de investigación se lleven a cabo tomando en cuenta los mejores intereses de la comunidad. Todos los colaboradores tienen responsabilidades específicas entre sí y en cuanto al proceso de investigación. Es más probable que la investigación se lleve a cabo con éxito si todos los colaboradores cuentan con la información y capacitación necesarias para cumplir con sus responsabilidades.")
24
Protecciones básicas a la persona
Principios Éticos Básicos se pueden garantizar A través de: Regulaciones que contienen tres protecciones básicas para sujetos humanos: Garantía institucional o representantes comunitarios Revisión del IRB Consentimiento informado

25
Garantía Institucional
Es la documentación de un compromiso institucional para cumplir con las regulaciones Éticas y mantener un adecuado programa y procedimientos para la protección de sujetos humanos El principal mecanismo de cumplimiento supervisado para la protección de humanos como sujetos de investigaciones

26
Garantía Institucional
Oficial institucional Es la persona autorizada para actuar en nombre de la institución Los oficiales institucionales no pueden autorizar o aprobar la conducción de investigaciones en sujetos humanos que no sean aprobadas por el IRB. Las investigaciones aprobadas por un IRB pueden ser sujetas de revisión y aprobación o desaprobación posterior por los oficiales de la institución.

28
Responsabilidades del investigador
El investigador tiene la responsabilidad fundamental de proteger a todas las personas que participan en la investigación y de colocar el bienestar de los participantes por encima de los intereses de la ciencia y la sociedad. El investigador debe considerar esta responsabilidad no sólo como un requisito regulador o jurídico, sino también como una exigencia para llevar a cabo la investigación de conformidad con las normas y los principios éticos universales. El investigador debe elaborar protocolos de investigación científica y técnicamente correctos basados en los mejores métodos científicos. Las Normas del Consejo de Organizaciones Internacionales de las Ciencias Médicas (CIOMS, por sus siglas en inglés) recomiendan que "el protocolo sea evaluado científica y éticamente por uno o más comités de ética apropiadamente constituidos". Es la obligación del investigador presentar el protocolo que sea revisado por un comité de ética reconocido. Además, el investigador debe asegurarse de que cada participante dé su consentimiento informado antes de ser admitido al estudio. Este consentimiento informado debe cumplir con los requisitos abarcados en la sección V. El investigador debe proteger la confidencialidad de los participantes, según los términos declarados en el consentimiento informado
 recomiendan que el protocolo sea evaluado científica y éticamente por uno o más comités de ética apropiadamente constituidos . Es la obligación del investigador presentar el protocolo que sea revisado por un comité de ética reconocido. Además, el investigador debe asegurarse de que cada participante dé su consentimiento informado antes de ser admitido al estudio. Este consentimiento informado debe cumplir con los requisitos abarcados en la sección V. El investigador debe proteger la confidencialidad de los participantes, según los términos declarados en el consentimiento informado.")
30
Responsabilidades del patrocinador
Por lo general, el patrocinador proporciona los fondos y otros recursos necesarios para realizar la investigación. Por tanto, los patrocinadores se encargan de brindar un ambiente que promueva la integridad, la objetividad y los estándares éticos más estrictos. Los patrocinadores pueden crear este ambiente de varias maneras: Garantizar la revisión, aprobación y supervisión apropiadas por parte de un comité de ética y un grupo de representantes comunitarios (donde se encuentre disponible). Colaborar exclusivamente con investigadores calificados y proporcionarles toda la capacitación necesaria para llevar a cabo la investigación en forma adecuada. Colaborar con los investigadores para formular políticas, procedimientos y normas por escrito antes de que se inicie la investigación. Monitorear la investigación, colaborar para garantizar que todos los datos sean auténticos, y procesados y analizados correctamente. Apoyar técnica y económicamente el establecimiento y el funcionamiento de un grupo apropiado de asesoría a la comunidad siempre que sea posible y necesario. Colaborar con colegas locales de investigación para garantizar la publicación y difusión de los resultados de la investigación.
. Colaborar exclusivamente con investigadores calificados y proporcionarles toda la capacitación necesaria para llevar a cabo la investigación en forma adecuada. Colaborar con los investigadores para formular políticas, procedimientos y normas por escrito antes de que se inicie la investigación. Monitorear la investigación, colaborar para garantizar que todos los datos sean auténticos, y procesados y analizados correctamente. Apoyar técnica y económicamente el establecimiento y el funcionamiento de un grupo apropiado de asesoría a la comunidad siempre que sea posible y necesario. Colaborar con colegas locales de investigación para garantizar la publicación y difusión de los resultados de la investigación.")
31
Responsabilidades del patrocinador en la investigación internacional
Todas las normas internacionales recomiendan que los patrocinadores externos brinden asistencia financiera, educativa y técnica para promover el desarrollo de capacidad en el área de la ética de la investigación, incluido el apoyo de un comité de ética independiente. Siempre que sea posible, el patrocinador debe obtener la revisión y aprobación de la investigación por parte de un comité de ética local y asegurarse de que la investigación propuesta cumpla con los requisitos éticos, reguladores y jurídicos, tanto a nivel local como nacional. Antes de iniciarse el estudio, los patrocinadores son responsables de hablar con sus colaboradores en la localidad sobre la relevancia de la investigación respecto a las necesidades y prioridades locales y sobre los posibles beneficios a las comunidades participantes. Una vez se haya concluido la investigación, particularmente en el caso de un resultado favorable como un producto autorizado eficaz, los patrocinadores deben hacer esfuerzos razonables para garantizar que los participantes o la comunidad en general puedan adquirir los productos de la investigación. Existe un acuerdo general de que los patrocinadores o los investigadores son responsables de brindar atención médica gratuita a los participantes cuando se presentan lesiones o complicaciones como resultado de la investigación. Cualquier limitación o condición debe indicarse en el consentimiento informado. En algunos estudios puede que sea necesario tomar en cuenta la responsabilidad de los patrocinadores de brindar otros tipos de atención médica y ésta debe ser especificada en el protocolo de investigación y en el formulario de consentimiento informado.

33
Protecciones básicas Principios Éticos Básicos se pueden garantizar
A través de: Regulaciones que contienen tres protecciones básicas para sujetos humanos: Garantía institucional Revisión del IRB

34
Comité Institucional de Revisión Ética (IRB)
El Comité Institucional de Revisión Ética (IRB) es un comité establecido para proteger los derechos y el bienestar de los sujetos humanos reclutados para participar en actividades de investigación conducida bajo los auspicios de la institución a la que esta afiliada.
 es un comité establecido para proteger los derechos y el bienestar de los sujetos humanos reclutados para participar en actividades de investigación conducida bajo los auspicios de la institución a la que esta afiliada.")
35
Comité Institucional de Revisión Ética (IRB)
Debe ser suficientemente calificado a través de la experiencia y el prestigio de sus miembros Sin importar su herencia racial, cultural Con actitudes que promuevan el respeto por sus opiniones y consejos en la salvaguarda de los derechos y bienestar de los sujetos humanos.

36
Comité Institucional de Revisión Ética (IRB) (Continuación)
Ser de carácter multidisciplinario. Los IRB’s deben tener por lo menos cinco Al menos un miembro del comité no debe ser del área científica. Al menos un miembro del comité debe ser estrictamente del área científica. Al menos un miembro del comité no debe estar afiliado con la institución. Deben considerarse ambos géneros.

37
Revisión del IRB La revisión del IRB tiene autoridad para aprobar, requerir modificaciones o desaprobar todas las investigaciones, Proponer cambios en investigaciones en sujetos humanos previamente aprobadas. El IRB conduce revisiones continuas de las investigaciones aprobadas en intervalos apropiados al grado de riesgo, pero no mas largos de una vez al año. El IRB tiene la autoridad de suspender o terminar investigaciones previamente aprobadas si no están siendo conducidas en concordancia con los requerimientos del IRB o si han sido asociadas con serios daños no esperados en los sujetos.

38
Comités de ética: Revisión de la investigación Para decidir si los estudios propuestos son éticos, el comité de ética debe tomar en cuenta seis temas básicos: El diseño científico y la ejecución del estudio. Al diseñar el estudio, el comité de ética debe pensar en la repercusión sobre la seguridad de los participantes. El reclutamiento de los participantes de la investigación. El comité de ética debe estudiar cómo reclutar a los participantes. Los factores comunitarios. El estudio debe tratar una necesidad o un problema local y debe diseñarse con una comprensión de la comunidad local. Para lograr esto, el comité de ética debe procurar incorporar las ideas y opiniones de los representantes comunitarios. La atención médica y la protección de los participantes de la investigación. El comité de ética debe observar qué tan positiva o negativamente afecta el estudio a los participantes o sus comunidades. El consentimiento informado. El comité de ética debe decidir si el formulario y el proceso de consentimiento son adecuados. Los representantes comunitarios pueden proporcionar una perspectiva importante sobre el proceso de consentimiento informado. Los aspectos de confidencialidad. El comité de ética debe revisar las medidas adoptadas por el equipo de estudio para proteger la confidencialidad de los participantes. En algunas investigaciones, el mayor riesgo que corren los participantes es el incumplimiento de su confidencialidad. Una vez que se hayan tratado estas inquietudes, el comité de ética puede otorgar su aprobación para dar inicio al estudio. Además, algunos comités de ética también revisan otros aspectos de la investigación, como la propiedad de los datos o los presupuestos.

41
Protecciones básicas Principios Éticos Básicos se pueden garantizar
A través: Regulaciones que contienen tres protecciones básicas para sujetos humanos: Garantía institucional Revisión del IRB Consentimiento informado

42
CONSENTIMIENTO INFORMADO
TEXTOS DE LA ETICA HIPOCRÁTICA “Haz todo con calma y orden ocultando al enfermo, durante tu actuación, la mayoría de las cosas. Dale las ordenes oportunas con amabili-dad y dulzura y distrae su atención; repréndele a veces estricta y severamente, pero otras anímale con solicitud y habilidad, sin mostrarle nada de lo que le va a pasar ni de su estado actual; pues muchos acuden a otros médicos por causa de esa declaración, antes mencionada, del pronóstico sobre su presente y su futuro”

43
CONSENTIMIENTO INFORMADO
DATOS HISTORICOS RELEVANTES Siglo XIX: Bajo la noción de los derechos individuales y colectivos surge el “Principio de Tolerancia” 1914: Schloendorff vrs Society of New York Hospitals (Sentencia del Juez Cardozo): “Todo ser humano de edad adulta y juicio sano tiene el derecho a determinar lo que debe hacerse con su propio cuerpo....” 1948: Código de Nuremberg plantea el consentimiento voluntario del sujeto 1964: Declaración de Helsinki
: Todo ser humano de edad adulta y juicio sano tiene el derecho a determinar lo que debe hacerse con su propio cuerpo : Código de Nuremberg plantea el consentimiento voluntario del sujeto. 1964: Declaración de Helsinki.")
44
CONSENTIMIENTO INFORMADO
DATOS HISTORICOS RELEVANTES Años 60-70: Movimiento de reinvindicación de los derechos civiles 1973: Promulgación de la “Primera Carta de Derechos de los Pacientes” por la Asociación Americana de Hospitales Década de lo 70: Desarrollo de la Bioética 1978: Informe Belmont (National Comission for the protection of Human Subjects of Biomedical and Behavioral Sciences) a petición del Congreso Norteamericano: Establece los principios éticos que deben orientar la investigación en seres humanos
 a petición del Congreso Norteamericano: Establece los principios éticos que deben orientar la investigación en seres humanos.")
45
CONSENTIMIENTO INFORMADO
Se refiere a la elección voluntaria de un individuo para participar en un acto médico (o investigación) basándose en una comprensión completa y profunda de sus propósitos , procedimientos, riesgos, beneficios, alternativas y cualquier otro factor que pueda afectar la decisión de participar
 basándose en una comprensión completa y profunda de sus propósitos , procedimientos, riesgos, beneficios, alternativas y cualquier otro factor que pueda afectar la decisión de participar.")
46
CONSENTIMIENTO INFORMADO
No es un simple evento ni es solo un formulario para ser firmado Es un proceso educativo que toma lugar entre el investigador y el participante Debe contener mínimo tres elementos: Información Comprensión Voluntario

47
REQUERIMIENTOS GENERALES
Debe ser obtenido del sujeto o representante legalmente autorizado La información debe ser dada en lenguaje entendible por el participante Darle suficiente oportunidad a los sujetos para decidir si quieren o no participar Debe ser dado sin coerción o influencia indebida No debe dar la impresión de pedirles la renuncia a sus derechos legales

49
COMPRENSION DEL CONSENTIMIENTO INFORMADO
Aunque se haya aprobado un procedimiento de CI, es la responsabilidad del médico (o investigador) asegurarse de que cada sujeto entienda la información, de lo contrario (NO DEBE SER INVOLUCRADOS). Se sugiere elaborar en algunos casos un cuestionario de comprensión del CI.
 asegurarse de que cada sujeto entienda la información, de lo contrario (NO DEBE SER INVOLUCRADOS). Se sugiere elaborar en algunos casos un cuestionario de comprensión del CI.")
50
COMPETENCIA CONSENTIMIENTO INFORMADO
Debe ser efectivo bajo las leyes del estado Solo pueden dar CI adultos legalmente competentes Solo los padres o tutores de menores de edad pueden dar el permiso respectivo Adultos con deficiencia de su desarrollo mental, ancianos con limitaciones cognitivas, personas inconscientes o semiinconscientes no pueden dar un CI La evaluación de competencia debe revisarse caso a caso

51
Una vez las personas comienzan a oír acerca de un nuevo estudio, el proceso de consentimiento informado está en marcha. Lo que las personas oyen a partir de este momento podría influir en su opinión del proyecto y su decisión de participar. Por este motivo, es importante que los representantes comunitarios ayuden a planear cómo lanzar un nuevo estudio

53
ELEMENTOS DEL CONSENTIMIENTO INFORMADO
Descripción de riesgos razonablemente previsibles Descripción de beneficios esperados Explicación de protección a la confidencialidad Explicación que la participación es voluntaria Consignar a quien contactar en caso de dudas Explicación de la política de compensación por daños Descripción de la investigación y participación de los sujetos incluyendo la identificación de procedimientos experimentales. Contactos para información o reclamos

54
RIESGOS Consignar todos los riesgos razonablemente previstos, incomodidades, inconveniencias y daños que puedan estar asociados con el acto médico ( o la actividad de investigación ) Si se identifica algún riesgo adicional no considerado al inicio, el proceso de CI y la documentación requerirán revisión para informar a los sujetos y estos deberán ser contactados de nuevo
 Si se identifica algún riesgo adicional no considerado al inicio, el proceso de CI y la documentación requerirán revisión para informar a los sujetos y estos deberán ser contactados de nuevo.")
55
BENEFICIOS Consignar cualquier beneficio que los sujetos puedan esperar razonablemente Ser francos sobre los beneficios y no sobre estimar o magnificar dichos beneficios En Investigación: Puede ser que el único beneficio sea el sentimiento de estar ayudando a la humanidad Debe ser muy cuidadoso al enunciarse pago a los sujetos de estudio o incentivos de tipo económico o en especies

56
CONFIDENCIALIDAD Deben indicar las personas u organizaciones que pueden tener acceso a la información. Grado de confidencialidad Uso futuro previsto de la información y de las muestras biológicas ejemplo: Auspiciadores, agencias financiadoras, agencias reguladoras, IRB o comités de ética

57
PARTICIPACION VOLUNTARIA ELEMENTOS ADICIONALES
La participación debe ser voluntaria Los sujetos pueden descontinuar su participación en cualquier momento No existe penalidad o pérdida de beneficios por no participar o descontinuar su participación ELEMENTOS ADICIONALES Riesgos no previstos Consecuencias de la salida del estudio Información de nuevos hallazgos Terminación de la participación Otros

58
COMPENSACION POR DAÑO Debe explicarse si se dará compensación por daños relacionados con la investigación (físico, psicológico, social, financiero o de otro tipo) Las regulaciones internacionales prohíben solicitarles a los sujetos o motivarlos a creer que renuncian a cualquiera de sus derechos legales El lenguaje que se utilice debe ser cuidadosamente seleccionado
 Las regulaciones internacionales prohíben solicitarles a los sujetos o motivarlos a creer que renuncian a cualquiera de sus derechos legales. El lenguaje que se utilice debe ser cuidadosamente seleccionado.")
59
CONTACTO DE PERSONAS Identificación de personas contacto para responder preguntas de la investigación y sus derechos como sujetos de ella Debe estar explícitamente establecido Una sola persona no es lo más adecuado ya que puede existir conflicto de intereses Debe tener al menos dos nombres de contactos con números de teléfonos y direcciones.

61
DOCUMENTACION DEL CONSENTIMIENTO INFORMADO
El CI es documentado por un formulario de comprensión aprobado por el IRB y firmado por el sujeto o su representante legal Debe entregársele una copia a la persona que firma el formulario Debe estar escrito en un lenguaje apropiado para su comprensión, evitando todo lenguaje técnico Se recomienda enfáticamente el uso de “primera persona” ejemplo: Yo he sido completamente informado acerca de.....

62
APLICACIÓN O NO DEL FORMULARIO DE CONSENTIMIENTO INFORMADO
El IRB puede autorizar a no aplicar el formulario en los siguientes casos: Cuando la investigación presenta un riesgo tan mínimo y envuelva procedimientos que no requieran consentimiento escrito Cuando la pérdida de la confidencialidad no implique ningún riesgo El hecho que un sujeto haya firmado el CI no significa que el/ella entiende que estuvo de acuerdo o que en realidad dio su consentimiento voluntario

63
BIBLIOGRAFIA Office for Human Research Protections. General Resource Links. Disponible en internet en Informed Consent Checklist. Disponible en internet en Informed Consent, Legally Effective and Prospectively Obtained. Disponible en internet en Informed Consent Non-English Speakers. Disponible en internet en of Confidentiality. Disponible en internet en U.S. Department of Health and Human Services. Informed Consent of Trial Subjects. Guidance for Industry. E6 Good Clinical Practice: Consolidated Guidance. P.17. April 1996. Human Subject Assurance Training. Investigator Responsabilities and informed Consent. Module 2. National Bioethics Advisory Commission. Temas sobre la ética y política en la investigación internacional: ensayos clínicos en los países en desarrollo. Resumen Ejecutivo. Bethesda, Maryland. Abril 2001 Currículo de Capacitación sobre Ética de la Investigación para los Representantes Comunitarios © Derechos de autor 2005, Family Health International (FHI)
")
64
GRACIAS POR SU ATENCION

Presentaciones similares


































>")










