Descargar la presentación
La descarga está en progreso. Por favor, espere

1
FARMACOCINETICA Universidad Nacional del Santa Facultad de Ciencias
Departamento Académico de Enfermería FARMACOCINETICA Ms. Vargas G. y Ms. Ramírez R. MS. D. VARGAS G.

2
Modificaciones que produce el sistema biológico al FARMACO.
FARMACOCINÉTICA Estudio cuantitativo de la relación concentración - tiempo. Cinética = Velocidad de pasaje del fármaco de un compartimento a otro. Espacio real o virtual del organismo al cual ingresa, se distribuye uniformemente y luego sale el fármaco (F) COMPARTIMENTO
 COMPARTIMENTO.")
3
Distribución uniforme
Entrada F Salida Distribución uniforme

5
Administración EV Administración ORAL Modelo monocompartimental
eliminación Modelo monocompartimental Modelo bicompartimental eliminación Administración ORAL eliminación

6
Modelo hidráulico ABSORCION ABSORCION Y ELIMNACION ELIMINACION
Tiempo (horas)
")
7
Sistema LADME ETAPAS FARMACOCINÉTICAS LIBERACIÓN ABSORCIÓN
DISTRIBUCIÓN METABOLISMO EXCRECIÓN CELULA BLANCO

8
datos de concentración sanguínea
BIODISPONIBILIDAD Cantidad de fármaco “disponible” en el sitio de acción o biofase. en un determinado tiempo (velocidad). datos de concentración sanguínea Evaluación a partir de RELACION DE FARMACODINAMIA CON FARMACOCINETICA Farmacodinamia Farmacocinética FARMACO PACIENTE
. datos de concentración sanguínea. Evaluación. a partir de. RELACION DE FARMACODINAMIA CON FARMACOCINETICA. Farmacodinamia. Farmacocinética. FARMACO. PACIENTE.")
9
CONSECUENCIA TERAPÉUTICA
determinación de concentraciones de fármaco en sangre, suero o plasma alternativa razonable Lugar difícilmente accesible... FASES DEL MEDICAMENTO FASE FARMACÉUTICA CONCENTRACIÓN DE DROGA EN BIOFASE FASE FARMACOCINÉTICA FASE FARMACODINÁMICA ACCIÓN CONSECUENCIA TERAPÉUTICA EFICACIA EFECTO INEFICACIA TOXICIDAD

10
VARIABILIDAD FARMACOCINETICA
Pediatría Obesos neonatos prematuros extremos Embarazo VARIABILIDAD FARMACOCINETICA Geriatría Pacientes críticos politraumáticos ventilación mecánica

11
EFICACIA TOXICIDAD CONCENTRACIÓN “La excesiva complejidad de los procesos fisiológicos y de los organismos en los que son observados impone respeto por el dicho de que no hay dos pacientes iguales” C. Bernard 1895

12
F F F F F- P Ma,i F Liberación Metabolismo M a Eliminación
FARMACOCINETICA VIA ORAL Liberación F Biotransformación Metabolismo Circulación general M a Eliminación Distribución F M i F F- P SITIO DE ACCIÓN RECEPTORES DEPOSITOS TISULARES Ma,i F

13
Esquema de la farmacocinética de los fármacos inhalados

14
MODELO DE FUNCIONAMIENTO DE FORMAS DE DOSIFICACIÓN ORAL
Medidas clínicas y/o PD Evaluación de la formulación Medidas de Farmacocinética 1er. Paso Formulación Fármaco en solución Pared intestinal Sangre Lugar de efecto Efecto Terapéutico Dosis ln Dosis RELACIÓN ENTRE DOSIS, CONCENTRACIÓN PLASMÁTICA Y EFECTO Intensidad del efecto farmacológico relacionado con la concentración de fármaco en plasma

15
Fármaco en Circulación Sistémica
LIBERACION DE FARMACOS ···· ····· Partículas Finas Desintegración Desagregación Gránulos o Agregados Forma de Dosificación Sólida Disolución Mayor Disolución Menor Disolución Mayor Fármaco en Solución (In vitro o in vivo) Forma de Dosificación Líquida Absorción in vivo Fármaco en Circulación Sistémica
 Forma de. Dosificación. Líquida. Absorción. in vivo. Fármaco en Circulación Sistémica.")
16
LIBERACIÓN CONTROLADA EN EL TIEMPO
ORIFICIO DE SALIDA DEL PRINCIPIO ACTIVO MEMBRANA SEMIPERMEABLE NUCLEO OSMOTICO CON PRINCIPIO ACTIVO

17
Forma de liberación controlada (Parche transdérmico)
")
18
Lugar de administración
Paso del fármaco circulación sistémica Inicio del efecto Intensidad del efecto

19
MECANISMOS DE ABSORCION DE FÁRMACOS POR MEMBRANAS CELULARES
O DIFUSIÓN PASIVA DIFUSIÓN FACILITADA FILTRACIÓN O DIFUSION 1 2 3 PINOCITOSIS PROTEINAS NO REQUIERE ENERGÍA REQUIERE ENERGÍA A favor de gradiente de concentración, estado de equilibrio en ambos lados de la membrana Contra gradiente concentración

20
TRANSPORTE ACTIVO ATP ADP CARACTERÍSTICAS DE LA ABSORCIÓN PASIVA
- Ley de difusión de Fick Liposolubilidad o coeficiente de partición lípido/agua - Gradiente de concentración a través de la membrana - Influencia del pH en procesos de absorción de fármacos: pKa de bases débiles y ácidos débiles > liposolubilidad mejor difusión a través de membranas TRANSPORTE ACTIVO ATP ADP

21
FACTORES DETERMINANTES DE LA ABSORCIÓN DE FÁRMACOS
CARACTERISTICAS DE UN FÁRMACO Tamaño y forma moleculares Solubilidad en el sitio de absorción Grado de ionización Liposolubilidad relativa de sus formas ionizada y no ionizada PROPIEDADES DE LA MEMBRANA Fluidez Flexibilidad Resistencia eléctrica Impermeabilidad relativa. PROTEINAS DE LA MEMBRANA

22
FACTORES FISICOQUIMICOS
Membranas celulares Procesos pasivos Electrolitos débiles y pH Transporte transmembrana

23
pH LOCAL DETERMINA IONIZACION DE LAS MOLÉCULAS
Moléculas de un mismo fármaco Ionizado No ionizado - neutra (carga eléctrica) (sin carga) + en equilibrio Depende de Sus propiedades químicas: Acido débiles Bases débiles pKa Del pH local del medio MEMBRANA CELULAR Ácidos débiles se ionizan en pH alcalino Bases débiles se ionizan en pH ácido pueden difundir no pueden difundir
 (sin carga) + en equilibrio. Depende. de. Sus propiedades químicas: Acido débiles. Bases débiles. pKa. Del pH local del medio. MEMBRANA CELULAR. Ácidos débiles se ionizan en pH alcalino. Bases débiles se ionizan en pH ácido. pueden. difundir. no pueden. difundir.")
24
SUPERFICIE DE ABSORCION
Propiedades fisicoquímicas del fármaco: Forma farmacéutica empleada Fármaco en solución Fármaco en forma sólida SOLUBILIDAD acuosa Mayor absorción oleosa Menor absorción Aun menor absorción CONCENTRACION DEL FARMACO A MAYOR CONCENTRACION DEL FARMACO MAYOR ABSORCION Lugar de absorción: A MAYOR SUPERFICIE MAYOR ABSORCION Ejm. Mucosa respiratoria y peritonel gran superficie de absorción A MAYOR FLUJO O CIRCULACION SANGUINEA MAYOR ABSORCION SUPERFICIE DE ABSORCION Flujo sanguíneo

25
ABSORCIÓN SEGÚN VÍA DE ADMINISTRACIÓN
Depende de: Endotelio de capilar VIA IM, SC Forma farmacéutica empleada. Hidrólisis del pH ácido del estómago Velocidad vaciado gástrico Solubilidad en lípidos (Difusión pasiva) Presencia de alimentos Concentración Tránsito intestinal Metabolismo de primer paso VIA ORAL Epitelio ● ● ● ● Medicamento ● ● Sangre ● ● ● ● ● ● ◦ ◦ ● ● ◦ ◦ ◦ ◦ ◦ ◦ ◦ ◦ ◦ ◦ ● ◦ ◦ ● ◦ ● ● ◦ ◦ ◦ ◦ ◦ ◦ ◦ ◦ ◦ ● ● Fármaco o PA ● ● Liquido intersticial ● ● Comprimidos o cápsulas con cubierta gastrorresistente o entérica ● Lumen intestinal ● ● ● Protege: fármacos que se alteran por jugo gástrico, se disgregan en intestino delgado mucosa gástrica de fármacos irritantes
 Presencia de alimentos. Concentración. Tránsito intestinal. Metabolismo de primer paso. VIA ORAL. Epitelio. ● ● ● ● Medicamento. ● ● Sangre. ● ● ● ● ● ● ◦ ◦ ● ● ◦ ◦ ◦ ◦ ◦ ◦ ◦ ◦ ◦ ◦ ● ◦ ◦ ● ◦ ● ● ◦ ◦ ◦ ◦ ◦ ◦ ◦ ◦ ◦ ● ● Fármaco. o PA. ● ● Liquido intersticial. ● ● Comprimidos o cápsulas con cubierta gastrorresistente o entérica. ● Lumen intestinal. ● ● ● Protege: fármacos que se alteran por jugo gástrico, se disgregan en intestino delgado. mucosa gástrica de fármacos irritantes.")
26
valor del pH corporal intestino delgado (con contenido) 7.5 - 8.1
intestino delgado (superficie absorbente) 5.3 saliva 6.4 orina (valor medio) orina (valor bajo) orina (valor alto) 8 sangre arterial 7.4 vagina intracelular jugo gástrico (ulcera gástrica) 1.5 jugo gástrico (normal) jugo gástrico (carcinoma de estomago) 5.7 liquido cefalorraquídeo 7.5 piel (superficie) 4.5 piel (interdigital, pliegue y afección cutánea)
 5.3. saliva orina (valor medio) orina (valor bajo) orina (valor alto) 8. sangre arterial vagina intracelular jugo gástrico (ulcera gástrica) 1.5. jugo gástrico (normal) jugo gástrico (carcinoma de estomago) 5.7. liquido cefalorraquídeo piel (superficie) 4.5. piel (interdigital, pliegue y afección cutánea)")
27
Absorción otras vías enterales
Vía sublingual Vía rectal Se evita el paso intestinal y hepático La sustancia debe estar en contacto con la mucosa Efecto rápido e intenso Alternativa a la vía oral Evita parcialmente metabolismo de primer paso MUCOSA NO PREPARADA SUPERFICIE DE ABSORCIÓN PEQUEÑA TIEMPO DE CONTACTO CORTO Errática en los niveles plasmáticos MUCOSAS Uso local: rinofaríngea, conjuntiva, uretral, vaginal Uso sistémico: vía nasal.

28
Absorción vías parenterales
Intravenosa Acción inmediata. Control exacto de la dosis. > RIESGOS MÉTODOS DE ADMINISTRACIÓN Directa Bolus (< 1ml/ min) o lenta (2-5 min). Infusión: Intermitente o corta (30 a 60 min) Continua (24 horas)
 o lenta (2-5 min). Infusión: Intermitente o corta (30 a 60 min) Continua (24 horas)")
29
Intramuscular Subcutánea
Fármacos no absorbibles por VO Absorción relativamente rápida (10-30 min): soluciones acuosas Preparados liberación prolongada (“depot”) Algunos fármacos precipitan al pH múscular Sitio de administración Subcutánea Autoadministración Absorción más lenta que VIM. Absorción, por lo general, completa Preparados depot DEPENDEN DEL FLUJO SANGUINEO
: soluciones acuosas. Preparados liberación prolongada ( depot ) Algunos fármacos precipitan al pH múscular. Sitio de administración. Subcutánea. Autoadministración. Absorción más lenta que VIM. Absorción, por lo general, completa. Preparados depot. DEPENDEN DEL. FLUJO SANGUINEO.")
30
VÍA RESPIRATORIA Aerosoles (líquidos/polvos): Efectos locales
Técnica empleada Tamaño de las partículas Gases y anestésicos volátiles: Absorción rápida

31
liberación controlada
Absorción Vía cutánea Resistencia a difusión pasiva Formas de liberación controlada Velocidad lenta Barrera lipófila cerrada Acción local Absorción sistémica

32
ABSORCIÓN Y VIAS DE ADMINISTRACIÓN
Vía Ventajas Desventajas Ejemplos ENTERAL Oral Fácil, segura, conveniente Absorción limitada o errática de algunas drogas; posibilidad de inactivación hepática Analgésicos, sedantes e hipnóticos, etcétera Sublingual Inicio rápido del efecto. No se inactiva en el hígado El fármaco debe absorberse en la mucosa oral Nitroglicerina Rectal Opción de la vía oral. Efectos locales en la mucosa rectal Absorción pobre o incompleta. Riesgo de irritación rectal Laxantes, supositorios y otros Inhalación Inicio rápido. Aplicación directa en alteraciones respiratorias. Gran superficie de absorción Riesgo de irritación tisular. Problemas de dosificación Anestésicos generales, agentes antiasmáticos Inyección (SC, IV, IP, intratecal*) Administración a órganos blanco. Inicio rápido Riesgo de infección. Dolor. Imposibilidad de recuperar la droga. Sólo fármacos solubles Insulina, antibióticos, drogas anticancerígenas, narcóticos Tópica Efectos locales sobre la superficie de la piel Sólo eficaz en capas superficiales de la piel Ungüentos, cremas, gotas nasales y oculares, preparaciones vaginales
 Administración a órganos blanco. Inicio rápido. Riesgo de infección. Dolor. Imposibilidad de recuperar la droga. Sólo fármacos solubles. Insulina, antibióticos, drogas anticancerígenas, narcóticos. Tópica. Efectos locales sobre la superficie de la piel. Sólo eficaz en capas superficiales de la piel. Ungüentos, cremas, gotas nasales y oculares, preparaciones vaginales.")
33
DISTRIBUCIÓN DEPOSITOS TISULARES Fármaco - Proteína Metabolitos
Movimiento del medicamento desde la sangre hacia los órganos en los va actuar y a los que lo van a eliminar .¿tejidos al que accede? ORGANOS DIANA DEPOSITOS TISULARES ¿A qué velocidad y en qué concentración? CIRCULACIÓN GENERAL Fármaco libre Fármaco - Proteína Metabolitos ABSORCIÓN EXCRECIÓN Comienzo del efecto Intensidad del efecto BIOTRANSFORMACIÓN

34
DISTRIBUCION DE FARMACO ADMINISTRADO POR DIFERENTES VIAS

35
FACTORES QUE AFECTAN LA DISTRIBUCIÓN
ESTADO DEL FARMACO EN LA SANGRE Libre Fijados a los eritrocitos Unidos a proteínas plasmáticas difusión. retrasa la eliminación. prolonga el efecto. favorece la absorción intestinal. FIJACIÓN EN TEJIDOS Antipalúdicos Tetraciclinas Griseofulvina Clorofenotano Tiopental FUERZAS DE UNIÓN Fármaco - proteína plasmática = Interacción fármaco -receptores Enlace covalente Enlace iónico Enlace dipolo- dipolo Enlace hidrógeno Combinación con grupos sulfidrilos Factores fisiológicos Gasto cardiaco Flujo sanguíneo corazón, riñones, hígado y encéfalo músculos, vísceras, piel y grasa pH sanguíneo

36
Unión a proteínas plasmáticas
... no se une a receptor, no causa efectos FRACCION UNIDA A ... no pueden ser eliminadas ● ● ● ● ● ● ● ● ... Se une a receptor causa efectos SANGRE FRACCION LIBRE ... Pueden ser eliminadas ● ● ● ● ● ● ● ● Proteína: ALBUMINA Características de unión a fármacos: Flujo sanguíneo regional Corazón Cerebro Hígado Riñones Por interacciones electrostáticas Diferencias entre los fármacos Reversible Saturable Fracción libre y fracción unida, en equilibrio Competitiva PM bajo, facilita filtración en capilar Liposolubilidad

37
CAPILAR MEMBRANA Hidrosoluble Liposoluble CELULAR PLASMA ESPACIO
INTRACELULAR Proteína combinada Hidrosoluble Liposoluble

38
PASO DE FÁRMACOS AL SISTEMA NERVIOSO CENTRAL
Barrera hematoencefálica Área postrema Eminencia media Glándula pineal Fármacos anticolinesterasicos (amonio cuaternario) Anticolinérgicos (buscapina y metilbromuro de escopolamina) L- Dopa ROTURA DE LA BARRERA HEMATOENCEFÁLICA Hipercapnia Convulsiones Infecciones virales y/o bacterianas Hipertensión Arterial Meningosis leucémica Diuréticos osmóticos
 Anticolinérgicos (buscapina y metilbromuro de escopolamina) L- Dopa. ROTURA DE LA BARRERA HEMATOENCEFÁLICA. Hipercapnia. Convulsiones. Infecciones virales y/o bacterianas. Hipertensión Arterial. Meningosis leucémica. Diuréticos osmóticos.")
39
Hematoencefalica - BHE
Barrera Hematoencefalica - BHE Cohesión membranas capilares DP: liposolubilidad Transporte activo

40
BARRERA HEMATOPLACENTARIA
Epitelio trofoblástico Tejido conjuntivo coriónico Endotelio capilar Barbitúricos, morfina, anestésicos locales Glucosa Aminoácidos y iones Inmunoglobulina Sustancias cuaternarias e hidrosolubles FARMACO Peso molecular Liposolubles Metaboliza Algunos fármacos se acumulan

41
SECUESTRO IÓNICO Por diferencias de pH entre dos compartimentos AH
H+ + A- Compartimento 1 Compartimento 2 Fármacos ácidos en medio con pH básico Fármacos básicos en medio con pH ácido

42
Depósitos de medicamentos
Lugar donde el fármaco puede acumularse y almacenarce Proteínas, grasa o calcio Reversible o irreversible Reversible: puede darse el fenómeno de redistribución Volumen de distribución Parámetro utilizado para poder expresar las características de distribución de un fármaco. Relaciona la cantidad de fármaco en el organismo con la concentración plasmática determinada a un tiempo dado. VD = Cantidad del fármaco en cuerpo (dosis)/C plasmática
/C plasmática.")
43
AGUA TOTAL DEL ORGANISMO
60% PESO CORPORAL V= 0.6 L/Kg VOLUMEN INTRACELULAR 40% V= 0.4 L/Kg VOLUMEN EXTRACELULAR 20% V= 0.2 L/Kg VOLUMEN INTERSTICIAL 15% V= 0.15 L/Kg VOLUMEN PLASMATICO 5% V= 0.05 L/Kg

44
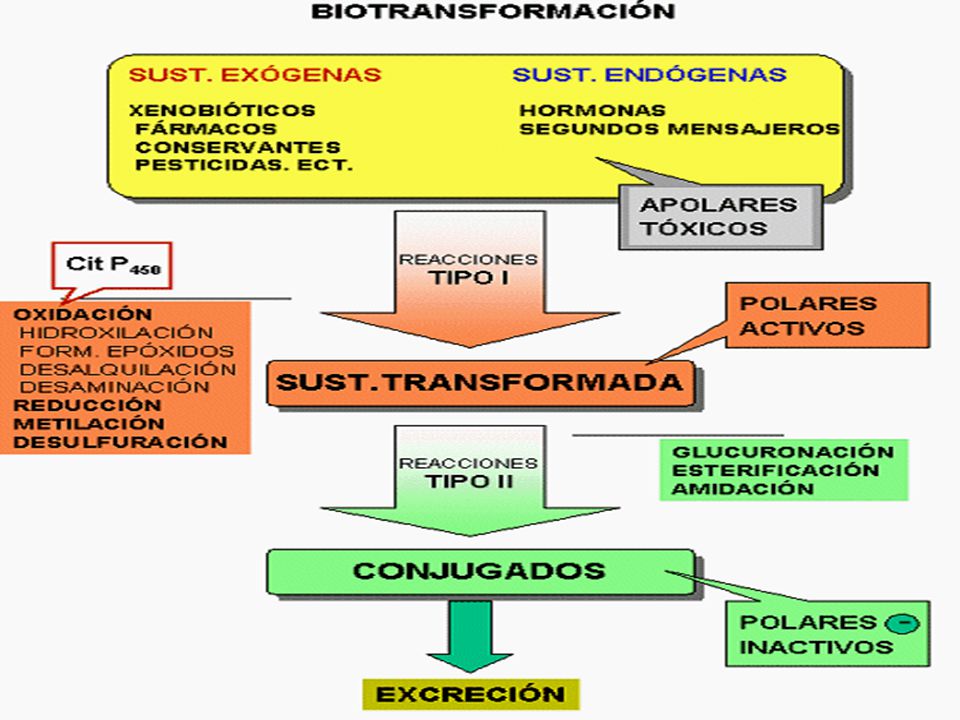
METABOLISMO BIOTRANSFORMACIÓN Hígado de Medicamento Tuvo digestivo
Modificación del fármaco en el organismo, se convierte en metabolitos más ionizados, más polares, más hidrosolubles, menos difusibles y más fácilmente eliminables que el fármaco BIOTRANSFORMACIÓN METABOLISMO de Medicamento en Hígado Tuvo digestivo inactivación por jugo gástrico: eritromicina y benicilpenicilina. hidrólisis de la insulina por proteasas hidrolasas (acetilcolina, succinilcolina, ác. acetilsalicílico) Plasma Riñón Pulmón nitritos
 Plasma. Riñón. Pulmón. nitritos.")
45
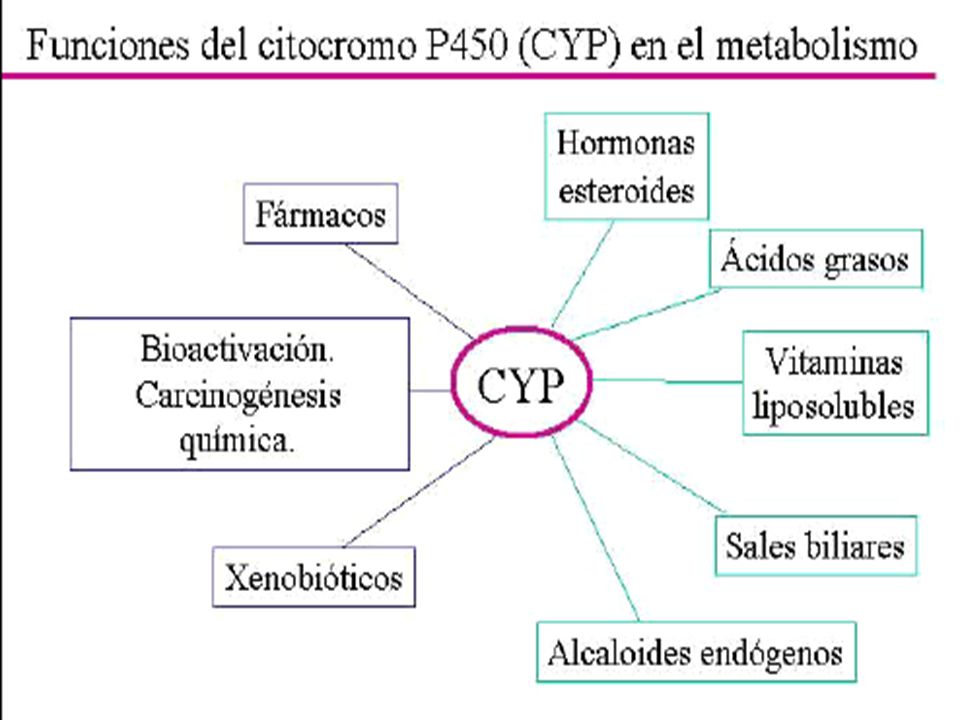
REACCIONES DE BIOTRANSFORMACIÓN:
Sistema microsomal Mitocondrias Enzimas solubles Lisosomas Microflora SISTEMA MICROSOMAL: hierro P450 ( hemo ) NADPH- citocromo P450 ( reductasa ) (flavina) ( flavina – adenina ) ( 1 – 2 electrones ) Monóxido de carbono
 NADPH- citocromo P450 ( reductasa ) (flavina) ( flavina – adenina ) ( 1 – 2 electrones ) Monóxido de carbono.")
48
CIRCULACIÓN ENTEROHEPÁTICA

49
EFECTO DE PRIMER PASO Dosis de Felodipino Biodisponibilidad Felodipino
Hígado Vena porta Intestino delgado Luz intestinal Enterocito Inhibidor Hepatocito Sinusoide

50
Actividad farmacológica: Mantener :
BIOTRANSFORMACIÓN: Bioactivación : Sulindaco reducción metabolito activo ( grupo sulfóxido ) Bioinactivación: Acetilcolina hidrolización ác. acético y colina ( Acetilcolinesterasa ) metabolitos inactivos Actividad farmacológica: Mantener : Diazepam N – Desmetildiazepam ( hrs. ) ( hrs. ) Modificar : Loxapina N-desmetilación amoxapina Toxicidad : Paracetamol N-hidroxilado
 Bioinactivación: Acetilcolina hidrolización ác. acético y colina. ( Acetilcolinesterasa ) metabolitos inactivos. Actividad farmacológica: Mantener : Diazepam N – Desmetildiazepam. ( hrs. ) ( hrs. ) Modificar : Loxapina N-desmetilación amoxapina. Toxicidad : Paracetamol N-hidroxilado.")
51
METABOLISMO HEPATICO Metabolitos Biotransformación Enzimática
salen por vena cava inferior y por bilis Biotransformación Enzimática

52
ELIMINACION Metabolitos Metabolitos activos NO POLAR inactivos ACTIVO
HIDROSOLUBLE Metabolitos Metabolitos activos ELIMINACION NO POLAR ACTIVO inactivos Profarmaco Fármaco Sin actividad farmacológica Con actividad farmacológica

53
metabolitos conjugados
FARMACO inactivos, en ocasiones: Metabolitos activos Metabolitos tóxicos Profármaco Fármaco metabolitos conjugados hidrosolubles de fácil excreción. Conjugación con Ácido glucurónico Glutamina Ácido acético etc. Ácido sulfúrico Glicocola

56
CONSECUENCIAS DE BIOTRANSFORMACIÓN ENZIMÁTICA DE FÁRMACOS
Facilita excreción renal del fármaco Inactivación del fármaco Incrementa la acción terapéutica (codeína morfina) Activación de profármacos Propicia toxicidad del fármaco.
 Activación de profármacos. Propicia toxicidad del fármaco.")
57
FACTORES QUE MODIFICAN EL METABOLISMO DE LOS FÁRMACOS
Químicos: D-estereoisómeros L-estereoisómero Farmacológicos: Vía de administración Dosis Unión a proteínas plasmáticas pH. urinario Inhibidores de la biotransformación Estimulantes de la biotransformación Patológicos: Estrés Cirrosis, metabolismo ↓

58
FACTORES FISIOLOGICOS
Presencia del P-450 Feto de 8 semanas Procesos de oxidación en el microsoma hepático Capacidad biotransformante del feto a lo largo de la vida intrauterina Enz. influida por agente estimulante o inhibidores Capacidad biotransformante inmadura RN Prematuro, > inmadurez metabólica. Enz. inducibles. EDAD Inmadurez renal, riesgo de intoxicación evidente Ej. kernicterus por insuficiente glucuronidación de bilirrubina Síndrome de niño gris por insuficiente glucuronidación del cloranfenicol < capacidad biotransformante, por: - de dotación enzimática hepática - reducción del flujo hepático. ANCIANO Reducción de función renal Ambos vida media biológica del fármaco y riesgo de acumulación tóxica.

59
SEXO El estado hormonal influye sobre la actividad de ciertas enzimas microsómicas, a las cuales estimulan o inhiben. Testosterona, vida media de antipirina, por estimulación de su metabolismo. Anabolizantes niveles de oxifenbutazona, por inhibición de glucuronidación Anticonceptivos orales inhiben metabolismo de antipirina, fenilbutazona Gestágenos, estimulan metabolismo de testosterona. en la actividad metabolizante de la fracción microsómica.

60
Genéticos: diferencia entre especies
diferencias dentro de una misma especie. inactivadores lentos (homocigóticos recesivos) inactivadores rápidos (homocigóticos dominantes) POLIMORFISMOS GENETICOS : Capacidad de cada persona para metabolizar un fármaco por una vía particular. Diferencias fenotípica Metabolismo deficiente ( ↑ incidencia de efectos adversos) Diferencias en la actividad metabólica (Recesivo autosómico)
 inactivadores rápidos (homocigóticos dominantes) POLIMORFISMOS GENETICOS : Capacidad de cada persona para metabolizar un fármaco por una vía particular. Diferencias fenotípica. Metabolismo deficiente ( ↑ incidencia de efectos adversos) Diferencias en la actividad metabólica (Recesivo autosómico)")
61
VARIABILIDAD INTERINDIVIDUAL
Fármaco Metabolitos Metabolitos Metabolitos Acetilador Acetilador Acetilador

62
Nutrición: Desnutrición Hidratos de carbono (P450 ↓ ) Hormonas:
Anemia hemolítica (sulfamidas, antipalúdicos de síntesis, etc.) Glucosa – 6 – fosfato deshidrogenasa ciclo de las pentosas NADPH Glutatión reducido integridad estructural del eritrocito Hormonas: Tx. H. Tiroideas Adrenalectomia (corticoides) Diabetes (↓ hexobarbital) Gestación Progesterona Factores ambientales (contaminantes)
 Glucosa – 6 – fosfato deshidrogenasa ciclo de las pentosas. NADPH. Glutatión reducido. integridad estructural del eritrocito. Hormonas: Tx. H. Tiroideas. Adrenalectomia (corticoides) Diabetes (↓ hexobarbital) Gestación. Progesterona. Factores ambientales (contaminantes)")
63
METABOLISMO DE LOS FÁRMACOS: INDUCCIÓN E INHIBICION ENZIMÁTICA
Inducción enzimática, Estimulación específica de síntesis de ciertos sistemas enzimáticos Enzimas con síntesis inducible: Monooxigenasas, Citocromo P-450, Glucuroniltransferasas Glutatión-transferasas. Inductores alteran expresión de Enz. Individuales: ↑ selectivo de capacidad de metabolizar fármacos, ↓disponibilidad del fármaco ↓ actividad del fármaco ↑ facilita destoxificación y eliminación. Ej. Fenobarbital usado como inductor Enz. en prematuros Glucocorticoides CYP3A4 Anticonvulsivos Isoniazida Acetona CYP2E1 Etanol

64
Inhibidores enzimáticos, inhiben síntesis de ciertos sistemas enzimáticos, incrementa la Cp. de los fármacos ENZIMAS DE niveles del fármaco original BIOTRANSFORMACIÓN prolongación de los efectos intrínsicos incidencia de intoxicaciones CYP2D INHIBIDA Quinidina Cimetidina Ketoconazol Inhiben metabolismo oxidativo agotamiento de los cofactores necesarios

65
pirogalol y tropolona catecol – O – metiltransferasa
catecolaminas acetilcolinesterasa acetilcolina alopurinol xantinooxidasa ácido úrico Inhibidores enzimáticos múltiples: SKF A (dietil-amino-difenil-propil-acetona) Lilly 18947; Sch 5705; Sch 5712, etc. (hexobarbital, petidina, anfetamina) Inhiben reacciones: destoxicación hidroxilación aromática desaminación oxidación de cadenas laterales SKF 525 A (inhibe las esterasas del plasma)
 Lilly 18947; Sch 5705; Sch 5712, etc. (hexobarbital, petidina, anfetamina) Inhiben reacciones: destoxicación hidroxilación aromática. desaminación. oxidación de cadenas laterales. SKF 525 A (inhibe las esterasas del plasma)")
66
Inhibidores de la biotransformación: catecolaminas
Inhibidores de la monoamino-oxidasa (enzimáticos múltiples) diamino-oxidasa guanidindesaminasa DOPA-descarboxilasa succinodeshidrogenasa etc. Inhibidores de la biotransformación: catecolaminas IMAO desaminación oxidativa indolaminas tono vital producen euforia Tiramina
 diamino-oxidasa. guanidindesaminasa. DOPA-descarboxilasa. succinodeshidrogenasa. etc. Inhibidores de la biotransformación: catecolaminas. IMAO desaminación oxidativa. indolaminas. tono vital. producen euforia. Tiramina.")
67
INTERACCIONES METABÓLICAS DE LOS FÁRMACOS: Beneficio terapéutico:
Hipertensión arterial ( Diuréticos más Beta-bloqueadores) Asma ( Corticoide más beta2 inhalados) Inmunodepresión ( azatioprina más ciclosporina)
 Asma. ( Corticoide más beta2 inhalados) Inmunodepresión. ( azatioprina más ciclosporina)")
68
Curso temporal de fármaco administrado por VO

69
(fácilmente excretable)
EXCRECIÓN DE FARMACOS Fármacos y sus metabolitos salen desde el torrente circulatorio al exterior del organismo FÁRMACO EN SANGRE VÍAS DE ELIMINACIÓN BIOTRANSFORMACIÓN ENZIMATICA EXCRECIÓN Saliva Sudor Renal Glándula mamaria Biliar Enterica Pulmonar Orina METABOLITO (fácilmente excretable) Aire expirado Leche materna Heces
 Aire. expirado. Leche. materna. Heces.")
70
VESICULA B. HIGADO RIÑON V V HECES ELIMINACION reabsorcion ORINA ORINA

71
EXCRECION RENAL Solo filtra la fracción libre Depende de:
Tubulo proximal SECRECION ACTIVA Transportadores Ácidos bases Desde tubulo distal REABSORCION PASIVA pKa del fármaco pH de la orina FILTRACION GLOMERULAR Solo filtra la fracción libre Depende de: Flujo sanguíneo PM del fármaco Mecanismo de transporte activo FARMACOS HIDROSOLUBLES ORINA

72
Secreción tubular: Transporte activo, diferenciado para ácidos y bases aniones orgánicos (penicilina, probenecida, salicilatos, ác. úrico) cationes orgánicos pasivo proximal del túbulo renal Posibilidad de competición por el mecanismo de transporte. Son independientes de la unión a proteínas Posibilidad de saturación

73
Reabsorción tubular orina Depende de: liposolublidad, ionización
pH de la orina Se puede modificar la excreción renal de fármacos varianado pH de orina; útil en tratamiento de intoxicaciones difusión pasiva Fármaco Liposoluble No ionizado luz tubular Sangre ● ● ● ● ● ● ● ● ● ● ● ● ● ● Fármaco hidrosoluble ionizado ● ● + ● ● + + + ● ● + ● ● + ● Orina tubular alcalina ác. Débiles Orina tubular ácida ác. débiles ● ● pH ● ● ● orina Orina tubular alcalina bases débiles Orina tubular ácida bases débiles Celulas tubulares Anfetaminas (acidificar)
")
75
EXCRECIÓN DE FÁRMACOS :
VOLUMEN DE EXCRECION URINARIA de un fármaco = resultado de la filtración glomerular y secreción tubular, menos la reabsorción tubular EXCRECIÓN DE FÁRMACOS : Fármacos liposolubles Filtran por riñón metabolizados Reabsorben metabolitos más polares fármacos hidrosolubles se excretan por riñón y bilis

76
EXCRECIÓN DE FÁRMACOS :
Características de eliminación de un fármaco: Elegir el fármaco adecuado duración del efecto numero de tomas deseadas valorar los factores que pueden alterarlas Velocidad con que se elimina tiempo que tarda en alcanzar y desaparecer su efecto Fluctuación de concentración plasmática Numero de tomas diarias

77
EXCRECIÓN DE FÁRMACOS :
pH urinario disociación reabsorción Intoxicación por ácidos débiles fenobarbital salicilatos alcalinizar sulfamidas, etc (fracción ionizada) Excreción biliar: Por secreción activa Sistemas diferentes para ácidos, bases y neutros. En membrana apical o canalicular Glucoproteínas P cationes organitos y proteínas MRP2 que eliminan aniones orgánicos En la membrana basal: Proteínas MRP aniones orgánicos a la sangre
 Excreción biliar: Por secreción activa. Sistemas diferentes para ácidos, bases y neutros. En membrana apical o canalicular. Glucoproteínas P cationes organitos y proteínas MRP2 que eliminan aniones orgánicos. En la membrana basal: Proteínas MRP1 aniones orgánicos a la sangre.")
78
Circulación enterohepática:
Se eliminan principalmente: Sustancias con elevado peso molecular Sustancias con grupos polares (cationes o aniones) que pueden ser del fármaco (amonio cuaternario) o radicales (glucuronatos o sulfatos) Compuestos no ionizables con una simetría de grupos lipófilos e hidrófilos ( digitoxina, digoxina y algunas hormonas) Algunos compuestos organometálicos Excreción intestinal Circulación enterohepática: Fármacos eliminados inalterados Reabsorberse pasivamente (gradiente de concentración) Los metabolitos pueden contribuir (flora intestinal) algunas bacterias contienen glucuronidasas
 que pueden ser del fármaco (amonio cuaternario) o radicales (glucuronatos o sulfatos) Compuestos no ionizables con una simetría de grupos lipófilos e hidrófilos ( digitoxina, digoxina y algunas hormonas) Algunos compuestos organometálicos. Excreción intestinal. Circulación enterohepática: Fármacos eliminados inalterados. Reabsorberse pasivamente (gradiente de concentración) Los metabolitos pueden contribuir (flora intestinal) algunas bacterias contienen glucuronidasas.")
79
EXCRECIÓN BILIAR sustancias polares y/o de elevado peso molecular transporte activo reabsorción intestinal de fármacos previamente excretados por bilis. Prolonga la permanencia de los fármacos en el organismo. Fármacos por VO.se excreta por heces sin absorción intestinal. Carbono activo.

80
Excreción por glándulas mamarias
Difusión pasiva. Pequeña [F] en leche pH leche ligeramente más ácido que el del plasma: Posibilidad que fármacos básicos queden secuestrados. Unión a proteínas o lípidos de la leche Acumulación de fármacos liposolubles, de tipo básico. Importancia para el recién nacido durante la lactancia.

81
OTRAS VÍAS DE EXCRECIÓN
Vía respiratoria: Vía rápida, poco frecuente, de importancia para anestésicos generales y sustancias volátiles Secreciones glandulares: Saliva sudor, lágrimas. Excreción de yoduros Piel y Faneras: Arsénico. Mercurio. Diagnóstico de intoxicaciones. Eliminación por diálisis: Características de fármacos diálizables y su utilidad

82
Aclaramiento hepático
Factores que influyen en la excreción renal: Edad: ↓ excreción renal en las edades extremas de la vida Interacciones farmacológicas: con agentes que modifiquen el pH o compitan con los sistemas de transporte Patologías concomitantes: Insuficiencia renal, prolonga la vida media de fármacos que se eliminan por vía renal.. ACLARAMIENTO Alcaramiento(Cl) Capacidad de une órgano para eliminar el fármaco. (ml. de plasma que el órgano aclara). Aclaramiento hepático Depende de: flujo sanguíneo hepático (QH), concentración de fármaco libre en sangre (Fls) y capacidad metabólica del hepatocito (aclaramiento intrínseco = Cli).
 Capacidad de une órgano para eliminar el fármaco. (ml. de plasma que el órgano aclara). Aclaramiento hepático. Depende de: flujo sanguíneo hepático (QH), concentración de fármaco libre en sangre (Fls) y capacidad metabólica del hepatocito (aclaramiento intrínseco = Cli).")
83
Aclaramiento renal del fármaco = creatinina filtración
si es mayor filtración y secreción tubular si es menor filtración y con reabsorción tubular Volumen de plasma que pasa por el riñón y es depurado de fármaco por unidad de tiempo creatinina < 1.5 mg/dl clearance creatinina ml/min Funcionamiento Normal 1.5 < creatinina < 2.5 mg/dl clearance creatinina ml/min leve insuf. Renal 2.5 < creatinina < 8 mg/dl clearance creatinina ml/min insuf. renal avanzada creatinina > 8 mg/dl clearance creatinina ml/min insuf. renal grave

84
ADMINISTRACION REPETIDAS

85
Cinética de eliminación orden uno
Velocidad del proceso es directamente proporcional a la concentración del fármaco Cp de fármaco exponencialmente con el Tiempo. La mayoría de fármacos utilizados en clínica, eliminados con cinética orden cero ...

86
Nivel plasmático (efecto)
Alcanzado cuando la dosis administrada reemplaza exactamente la cantidad eliminada de la dosis anterior Fármacos de cinética de eliminación orden uno, con dosificación fija Estado de equilibrio alcanzado luego de 4-5 veces t1/2 * Si t1/2 de un fármaco es larga se tardar algún tiempo en alcanzar la concentración terapéutica. ESTADO DE EQUILIBRIO Dosis Misma dosis a intervalos fijos Nivel plasmático (efecto) Nivel medio en sangre
 Nivel medio. en sangre.")
87
CRITERIOS DE SELECCIÓN DE PRINCIPIOS ACTIVOS QUE DEBEN MONITOREARSE EN SANGRE
El efecto farmacológico (terapéutico o tóxico) debe correlacionarese con las concentraciones séricas. Si el principio no se cumple, la medición de las concentraciones séricas no tienen significado. 2. Principios activos con ventana terapéutica estrecha. 3. Cuando no existe correlación entre la DOSIS y el efecto farmacológico. 4. Cuando el principio activo NO posee cinética de Orden 1 (proporcional).
 debe. correlacionarese con las concentraciones séricas. Si el principio no se cumple, la medición de las. concentraciones séricas no tienen significado. 2. Principios activos con ventana terapéutica estrecha. 3. Cuando no existe correlación entre la DOSIS y el efecto. farmacológico. 4. Cuando el principio activo NO posee cinética de Orden 1. (proporcional).")
88
INTERACCIONES METABÓLICAS DE LOS FÁRMACOS
Acción que un fármaco ejerce sobre otro, de modo que este experimenta un cambio cuantitativo o cualitativo en sus efectos. Absorción Dos o más Unión a proteínas fármacos Excreción por orina Biotransformación Tipos de interacciones: De carácter farmacéutico: Incompatibilidad de tipo físico – químico (mezcla) De carácter farmacocinéico: Modificaciones producidas por el fármaco desencadenante sobre los procesos de absorción, distribución y eliminación.
 De carácter farmacocinéico: Modificaciones producidas por el fármaco desencadenante sobre los procesos de absorción, distribución y eliminación.")
89
Tipos de interacciones:
De carácter farmacodinámico: Modificaciones en la respuesta, dando origen a fenómenos de Sinergia, antagonismo y potenciación En los receptores farmacológicos ( hipersencibilización y desensibilización de receptores, antagonismo y agonismo parcial) En procesos moleculares subsiguiente a la activación de receptores. En sistemas fisiológicos distintos < rapidez del metabolismo vía afectada (mecanismo principal de eliminación ) ↑ concentración plasmática prolonga o intensifica sus efectos intrínsicos F 1 F 2 ENZIMA
 En procesos moleculares subsiguiente a la activación de receptores. En sistemas fisiológicos distintos. < rapidez del metabolismo. vía afectada (mecanismo principal de eliminación ) ↑ concentración plasmática. prolonga o intensifica sus efectos intrínsicos. F 1. F 2. ENZIMA.")
90
INTERACCIONES

Presentaciones similares

















.>")
 Lo que el cuerpo le hace al fármaco Terminología Droga: Mezcla de.>")




