Descargar la presentación
La descarga está en progreso. Por favor, espere

1
Introducción a Bioperl
Verónica Jiménez Jacinto ibt.unam.mx Enero 2010

2
¿Qué es Bioperl? Bioperl es un esfuerzo comunitario para producir código en Perl, el cual sea útil, produciendo aplicaciones para la bioinformática, genómica y ciencias biológicas en general. Es un proyecto de software libre y fue fundamental en el proyecto de secuenciación del genoma humano. Consiste en un conjunto de módulos que facilita el desarrollo en Perl de herramientas bioinformáticas.

3
¿Por qué es atractivo BioPerl?
Paradigma de programación orientada a objetos. Clase Atributos Metodos Objeto Se base sobre la definición de clases en Perl que permiten generar objetos representando entidades biológicas, que después pueden interactuar entre ellas. Por ejemplo: Un objeto Aligment es generado a partir de objetos sequence, un objeto sequence tiene acceso a los objetos Annotation, SeqFeature y a bases de datos. Etc.

6
Instalación http://www.activestate.com/activeperl/
Por medio de la aplicación 'Sistema -> Administración -> gestor Synaptic', en realidad la manera sencilla de instalar cualquier software, Lo mismo se puede lograr desde la Terminal, por medio de: $ sudo apt-get install 'nombre:software' . Usando BIOPERL BUNDLE Instala Bioperl usando CPAN.: >perl -MCPAN -e "install Bundle::BioPerl" Otra manera; >perl -MCPAN -e shell cpan >install Bundle::BioPerl Instalando BIOPERL usando el shell de CPAN : >perl -MCPAN -e shell Then find the name of the Bioperl version you want: cpan>d /bioperl/ Ahora instala: cpan>install B/BI/BIRNEY/bioperl-1.4.tar.gz Instalando BIOPERL usando 'make' Descarga, descomprime y desempaqueta el archivo: >gunzip bioperl-1.2.tar.gz >tar xvf bioperl-1.2.tar >cd bioperl-1.2 Luego usa el comando make: >perl Makefile.PL >make >make test Para windows, se puede descargar una version en : Advanced Packaging Tool (Herramienta Avanzada de Empaquetado), abreviado APT, es un sistema de gestión de paquetes creado por el proyecto Debian. APT simplifica en gran medida la instalación y eliminación de programas en los sistemas GNU/Linux. No existe un programa apt en sí mismo, sino que APT es una biblioteca de funciones C++ que se emplea por varios programas de línea de comandos para distribuir paquetes. En especial, apt-get y apt-cache. Existen también programas que proporcionan un frontispicio para APT, generalmente basados en apt-get, como aptitude con una interfaz de texto ncurses, Synaptic con una interfaz gráfica GTK+, o Adept con una interfaz gráfica Qt. Existe un repositorio central con más de paquetes apt utilizados por apt-get y programas derivados para descargar e instalar aplicaciones directamente desde Internet, conocida como una de las mejores cualidades de Debian. o THE BIOPERL BUNDLE You typically need root privileges to install using CPAN. If you don't have these privileges please see INSTALLING BIOPERL IN A PERSONAL MODULE AREA for additional information. Install the Bioperl Bundle using CPAN. One way: >perl -MCPAN -e "install Bundle::BioPerl" Another way: >perl -MCPAN -e shell cpan>install Bundle::BioPerl o INSTALLING BIOPERL THE EASY WAY USING CPAN You can use the CPAN shell to install Bioperl. For example: >perl -MCPAN -e shell Then find the name of the Bioperl version you want: cpan>d /bioperl/ CPAN: Storable loaded ok Going to read /home/bosborne/.cpan/Metadata Database was generated on Tue, 24 Feb :55:23 GMT Distribution B/BI/BIRNEY/bioperl-1.2.tar.gz Distribution B/BI/BIRNEY/bioperl-1.4.tar.gz Now install: cpan>install B/BI/BIRNEY/bioperl-1.4.tar.gz If you've installed everything perfectly and all the network connections are working then you may pass all the tests run in the 'make test' phase. It's also possible that you may fail some tests. Possible explanations: problems with local Perl installation, network problems, previously undetected bug in Bioperl, flawed test script, problems with CGI script using for sequence retrieval at public database, and so on. Remember that there are over 700 modules in Bioperl and the test suite is running almost 9000 individual tests, a few failed tests may not affect your usage of Bioperl. If you decide that the failed tests will not affect how you intend to use Bioperl and you'd like to install anyway do: cpan>force install B/BI/BIRNEY/bioperl-1.4.tar.gz This is what most experienced Bioperl users would do. However, if you're concerned about a failed test and need assistance or advice then contact o INSTALLING BIOPERL THE EASY WAY USING 'make' The advantage of this approach is it's stepwise, so it's easy to stop and analyze in case of any problem. Download, then unpack the tar file. For example: >gunzip bioperl-1.2.tar.gz >tar xvf bioperl-1.2.tar >cd bioperl-1.2 Now issue the make commands: >perl Makefile.PL >make >make test If you've installed everything perfectly and all the network connections are working then you may pass all the tests run in the 'make test' phase. It's also possible that you may fail some tests. Possible explanations: problems with local Perl installation, network problems, previously undetected bug in Bioperl, flawed test script, problems with CGI script using for sequence retrieval at public database, and so on. Remember that there are over 700 modules in Bioperl and the test suite is running almost 9000 individual tests, a few failed tests may not affect your usage of Bioperl. If you decide that the failed tests will not affect how you intend to use Bioperl and you'd like to install anyway do: >make install
, abreviado APT, es un sistema de gestión de paquetes creado por el proyecto Debian. APT simplifica en gran medida la instalación y eliminación de programas en los sistemas GNU/Linux. No existe un programa apt en sí mismo, sino que APT es una biblioteca de funciones C++ que se emplea por varios programas de línea de comandos para distribuir paquetes. En especial, apt-get y apt-cache. Existen también programas que proporcionan un frontispicio para APT, generalmente basados en apt-get, como aptitude con una interfaz de texto ncurses, Synaptic con una interfaz gráfica GTK+, o Adept con una interfaz gráfica Qt. Existe un repositorio central con más de paquetes apt utilizados por apt-get y programas derivados para descargar e instalar aplicaciones directamente desde Internet, conocida como una de las mejores cualidades de Debian. o THE BIOPERL BUNDLE You typically need root privileges to install using CPAN. If you don t have these privileges please see INSTALLING BIOPERL IN A PERSONAL MODULE AREA for additional information. Install the Bioperl Bundle using CPAN. One way: >perl -MCPAN -e install Bundle::BioPerl Another way: >perl -MCPAN -e shell cpan>install Bundle::BioPerl o INSTALLING BIOPERL THE EASY WAY USING CPAN You can use the CPAN shell to install Bioperl. For example: >perl -MCPAN -e shell Then find the name of the Bioperl version you want: cpan>d /bioperl/ CPAN: Storable loaded ok Going to read /home/bosborne/.cpan/Metadata Database was generated on Tue, 24 Feb :55:23 GMT Distribution B/BI/BIRNEY/bioperl-1.2.tar.gz Distribution B/BI/BIRNEY/bioperl-1.4.tar.gz Now install: cpan>install B/BI/BIRNEY/bioperl-1.4.tar.gz If you ve installed everything perfectly and all the network connections are working then you may pass all the tests run in the make test phase. It s also possible that you may fail some tests. Possible explanations: problems with local Perl installation, network problems, previously undetected bug in Bioperl, flawed test script, problems with CGI script using for sequence retrieval at public database, and so on. Remember that there are over 700 modules in Bioperl and the test suite is running almost 9000 individual tests, a few failed tests may not affect your usage of Bioperl. If you decide that the failed tests will not affect how you intend to use Bioperl and you d like to install anyway do: cpan>force install B/BI/BIRNEY/bioperl-1.4.tar.gz This is what most experienced Bioperl users would do. However, if you re concerned about a failed test and need assistance or advice then contact o INSTALLING BIOPERL THE EASY WAY USING make The advantage of this approach is it s stepwise, so it s easy to stop and analyze in case of any problem. Download, then unpack the tar file. For example: >gunzip bioperl-1.2.tar.gz >tar xvf bioperl-1.2.tar >cd bioperl-1.2 Now issue the make commands: >perl Makefile.PL >make >make test If you ve installed everything perfectly and all the network connections are working then you may pass all the tests run in the make test phase. It s also possible that you may fail some tests. Possible explanations: problems with local Perl installation, network problems, previously undetected bug in Bioperl, flawed test script, problems with CGI script using for sequence retrieval at public database, and so on. Remember that there are over 700 modules in Bioperl and the test suite is running almost 9000 individual tests, a few failed tests may not affect your usage of Bioperl. If you decide that the failed tests will not affect how you intend to use Bioperl and you d like to install anyway do: >make install.")
7
¿Qué se puede hacer con Bioperl?
Acceder a secuencias locales o remotas Transformar formatos de diferentes Bases de datos Manipular secuencias individuales Buscar secuencias similares Crear y manipular alineamientos de secuencias Buscar genes y estructuras geonómicas sobre DNA Desarrollar código para leer anotaciones

8
Que se puede hacer con bioperl?
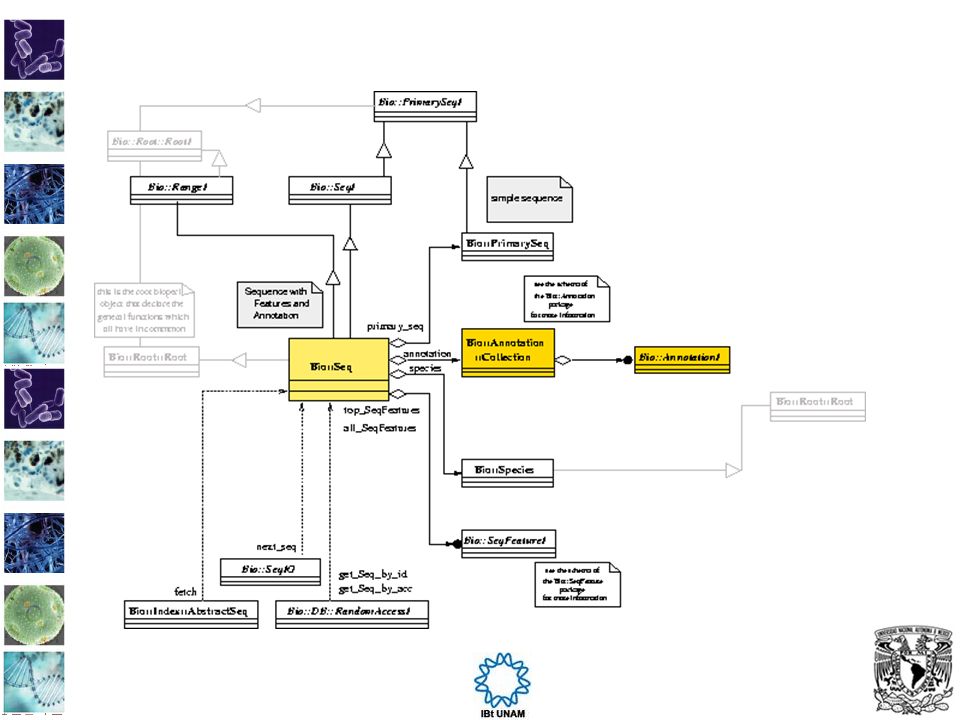
Sequences Bio::Seq es el principal objeto de la clase secuencia de Bioperl. Bio::PrimarySeq es un objeto secuencia sin características Bio::SeqIO Proporciona funciones para leer y escribir en secuencias desde archivos Bio::Tools::SeqStats proporciona estadísticas sobre secuencias. Bio::LiveSeq::* maneja cambio de secuencias. Bio::Seq::LargeSeq proporciona soporte para manejar secuencias muy grandes.

9
# this script will only work with an internet connection
Seq es el objeto cetral para manipular secuencias. Es una secuencia con características. use Bio::Perl; # this script will only work with an internet connection # on the computer it is run on $seq_object = get_sequence('swissprot',"ROA1_HUMAN"); my $seq_object2 = get_sequence('embl',"AI129902"); my $seq_object3 = get_sequence('genbank',"AI129902"); write_sequence(">roa1.fasta",'fasta',$seq_object2); cat roa1.fasta >unknown id qc41b07.x1 Soares_pregnant_uterus_NbHPU Homo sapiens cDNA clone IMAGE: ' similar to SW:ROA1_ SCHAM P21522 HETEROGENEOUS NUCLEAR RIBONUCLEOPROTEIN A1, A2/B1 HOMOLOG. ;contains MSR1.b2 MSR1 repetitive elemen t ;, mRNA sequence. CTCCGCGCCAACTCCCCCCACCCCCCCCCCACACCCC get_secuencias.pl EXCEPTION MSG: swissprot stream with no ID. Not swissprot in my book STACK Bio::SeqIO::swiss::next_seq /usr/local/lib/perl5/site_perl/5.8.0/Bio/SeqIO/swiss.pm:180 STACK Bio::DB::WebDBSeqI::get_Seq_by_acc /usr/local/lib/perl5/site_perl/5.8.0/Bio/DB/WebDBSeqI.pm:176 STACK Bio::Perl::get_sequence /usr/local/lib/perl5/site_perl/5.8.0/Bio/Perl.pm:510 STACK toplevel get_secuencias2.pl:2
; my $seq_object2 = get_sequence( embl , AI ); my $seq_object3 = get_sequence( genbank , AI ); write_sequence( >roa1.fasta , fasta ,$seq_object2); cat roa1.fasta. >unknown id qc41b07.x1 Soares_pregnant_uterus_NbHPU Homo sapiens cDNA clone IMAGE: similar to SW:ROA1_. SCHAM P21522 HETEROGENEOUS NUCLEAR RIBONUCLEOPROTEIN A1, A2/B1 HOMOLOG. ;contains MSR1.b2 MSR1 repetitive elemen. t ;, mRNA sequence. CTCCGCGCCAACTCCCCCCACCCCCCCCCCACACCCC. get_secuencias.pl EXCEPTION MSG: swissprot stream with no ID. Not swissprot in my book. STACK Bio::SeqIO::swiss::next_seq /usr/local/lib/perl5/site_perl/5.8.0/Bio/SeqIO/swiss.pm:180. STACK Bio::DB::WebDBSeqI::get_Seq_by_acc /usr/local/lib/perl5/site_perl/5.8.0/Bio/DB/WebDBSeqI.pm:176. STACK Bio::Perl::get_sequence /usr/local/lib/perl5/site_perl/5.8.0/Bio/Perl.pm:510. STACK toplevel get_secuencias2.pl:")
10
use Bio::Perl; # this script will only work with an # Internet connection # on the computer it is run on $seq_object = get_sequence('swissprot',"ROA1_HUMAN"); #uses the default database -nr in this case $blast_result = blast_sequence($seq_object); write_blast(">roa1.blast",$blast_result);
; #uses the default database -nr in this case. $blast_result = blast_sequence($seq_object); write_blast( >roa1.blast ,$blast_result);")
11
# gets a sequence from a file
$seqio = Bio::SeqIO->new( '-format' => 'embl' , -file => 'myfile.dat'); $seqobj = $seqio->next_seq(); # get from database $db = Bio::DB::swiss->new(); $seqobj = $db->get_Seq_by_acc('ROA1_HUMAN'); # make from strings in script $seqobj = Bio::Seq->new( -display_id => 'my_id', -seq => $sequence_as_string);
; $seqobj = $seqio->next_seq(); # get from database. $db = Bio::DB::swiss->new(); $seqobj = $db->get_Seq_by_acc( ROA1_HUMAN ); # make from strings in script. $seqobj = Bio::Seq->new( -display_id => my_id , -seq => $sequence_as_string);")
12
$seqobj = new_sequence("ATTGGTTTGGGGACCCAATTTGTGTGTTATATGTA",
“un nombre", "AL12232"); $seq_stats = Bio::Tools::SeqStats->new(-seq=>$seqobj); $monomer_ref = $seq_stats->count_monomers(); $codon_ref = $seq_stats->count_codons(); $weight = $seq_stats->get_mol_wt($seqobj);
; $seq_stats = Bio::Tools::SeqStats->new(-seq=>$seqobj); $monomer_ref = $seq_stats->count_monomers(); $codon_ref = $seq_stats->count_codons(); $weight = $seq_stats->get_mol_wt($seqobj);")
13
# gets sequence as a string from sequence object
$seqstr = $seqobj->seq(); # actual sequence as a string $seqstr = $seqobj->subseq(10,50); # slice in biological coordinates # retrieves information from the sequence # features must implement Bio::SeqFeatureI interface @features = $seqobj->get_SeqFeatures(); # just top level foreach my $feat ) { print "Feature ",$feat->primary_tag," starts ",$feat->start," ends ", $feat->end," strand ",$feat->strand,"\n"; # features retain link to underlying sequence object print "Feature sequence is ",$feat->seq->seq(),"\n" } # sequences may have a species if( defined $seq->species ) { print "Sequence is from ",$species->binomial_name," [",$species->common_name,"]\n"; # annotation objects are Bio::AnnotationCollectionI's $ann = $seqobj->annotation(); # annotation object # references is one type of annotations to get. Also get # comment and dblink. Look at Bio::AnnotationCollection for # more information foreach my $ref ( $ann->get_Annotations('reference') ) { print "Reference ",$ref->title,"\n"; # you can get truncations, translations and reverse complements, these # all give back Bio::Seq objects themselves, though currently with no # features transfered my $trunc = $seqobj->trunc(100,200); my $rev = $seqobj->revcom(); # there are many options to translate - check out the docs my $trans = $seqobj->translate(); # these functions can be chained together my $trans_trunc_rev = $seqobj->trunc(100,200)->revcom->translate();
; # actual sequence as a string. $seqstr = $seqobj->subseq(10,50); # slice in biological coordinates. # retrieves information from the sequence. # features must implement Bio::SeqFeatureI = $seqobj->get_SeqFeatures(); # just top level. foreach my $feat ) { print Feature ,$feat->primary_tag, starts ,$feat->start, ends , $feat->end, strand ,$feat->strand, \n ; # features retain link to underlying sequence object. print Feature sequence is ,$feat->seq->seq(), \n } # sequences may have a species. if( defined $seq->species ) { print Sequence is from ,$species->binomial_name, [ ,$species->common_name, ]\n ; # annotation objects are Bio::AnnotationCollectionI s. $ann = $seqobj->annotation(); # annotation object. # references is one type of annotations to get. Also get # comment and dblink. Look at Bio::AnnotationCollection for. # more information. foreach my $ref ( $ann->get_Annotations( reference ) ) { print Reference ,$ref->title, \n ; # you can get truncations, translations and reverse complements, these. # all give back Bio::Seq objects themselves, though currently with no. # features transfered. my $trunc = $seqobj->trunc(100,200); my $rev = $seqobj->revcom(); # there are many options to translate - check out the docs. my $trans = $seqobj->translate(); # these functions can be chained together. my $trans_trunc_rev = $seqobj->trunc(100,200)->revcom->translate();")
14
¿Qué se puede hacer en bioperl?
Databases Bio::DB::GenBank proporciona acceso a GenBank Bio::Tools::Run::StandAloneBlast corre BLAST localmente. Bio::Tools::Run::RemoteBlast corre BLAST remotamente. Bio::Tools::BPlite parsea un reporte BLAST Bio::Tools::BPpsilite parsea un reporte psiblast Bio::Tools::HMMER::Results parsea resultados de Cadenas de Markov.

15
Alignments Bio::SimpleAlign manipula y despliega alineamientos de múltiples secuencias Bio::LocatableSeq Son objetos secuencias con puntos de inicio y final para su localización relativa a otras secuencias o alineamientos. Bio::Tools::pSW alinea dos secuencias con el algoritmo Smith-Waterman. Bio::AlignIO Alinea dos secuencias con el algoritmo blast Bio::Clustalw es una interface del paquete Clustalw. Bio::TCoffee es una interface del paquete Tcoffee Bio::Variation::Allele maneja conjuntos de allelos. Bio::Variation::SeqDiff maneja conjuntos de mutaciones y variantes

16
Features and genes on sequences
Bio::SeqFeature es un objeto con las caracteristicas de una secuencia en Bioperl. Bio::Tools::RestrictionEnzyme localiza sitios de restriccion sitios en secuencias Bio::Tools::Sigcleave Encuentra sitios de corte en aminoacidos. Bio::Tools::OddCodes Rescribe secuencias de aminoacidos con codigos abreviados para especificar analisis estadisticos. (e.g., a hydrophobic/hydrophilic two-letter alphabet). Bio::Tools::SeqPattern Proporciona soporte para encontrar secuencias de patrones. Bio::LocationI proporciona una interface para localizar información de una seucencia Bio::Location::Simple maneja información de la localización de una secuencia, como una simple localización y como un rango. . Bio::Location::Fuzzy proporciona información de la localización que puede ser inexacta. Bio::Tools::Genscan es una interface para encontrar genes con el progrma Genscan Bio::Tools::Sim4::Results (and Exon) es una interface para encontrar exones de genes con el programa Sim4
. Bio::Tools::SeqPattern Proporciona soporte para encontrar secuencias de patrones. Bio::LocationI proporciona una interface para localizar información de una seucencia. Bio::Location::Simple maneja información de la localización de una secuencia, como una simple localización y como un rango. . Bio::Location::Fuzzy proporciona información de la localización que puede ser inexacta. Bio::Tools::Genscan es una interface para encontrar genes con el progrma Genscan. Bio::Tools::Sim4::Results (and Exon) es una interface para encontrar exones de genes con el programa Sim4.")
17
/product="pantoate beta alanine ligase protein"
>more $RHIZO_PUB/RE1PF/CP gbk LOCUS CP bp DNA circular BCT 10-MAR-2006 DEFINITION Rhizobium etli CFN 42 plasmid p42f, complete sequence. ACCESSION CP000138 VERSION CP GI: KEYWORDS . SOURCE Rhizobium etli CFN 42 REFERENCE 1 (bases 1 to ) AUTHORS Gonzalez,V., Santamaria,R.I., Bustos,P., Hernandez-Gonzalez,I., Medrano-Soto,A., Moreno-Hagelsieb,G., Janga,S.C., Ramirez,M.A., Jimenez-Jacinto,V., Collado-Vides,J. and Davila,G. TITLE The partitioned Rhizobium etli genome: Genetic and metabolic redundancy in seven interacting replicons … FEATURES Location/Qualifiers source /organism="Rhizobium etli CFN 42" /mol_type="genomic DNA" /strain="CFN 42" /db_xref="taxon:347834" /plasmid="p42f" promoter /note="sigma54 panCp promoter; Putative transcription initiation." gene /gene="panC" /locus_tag="RHE_PF00001" CDS /EC_number=" " /product="pantoate beta alanine ligase protein"
 AUTHORS Gonzalez,V., Santamaria,R.I., Bustos,P., Hernandez-Gonzalez,I., Medrano-Soto,A., Moreno-Hagelsieb,G., Janga,S.C., Ramirez,M.A., Jimenez-Jacinto,V., Collado-Vides,J. and Davila,G. TITLE The partitioned Rhizobium etli genome: Genetic and metabolic. redundancy in seven interacting replicons. … FEATURES Location/Qualifiers. source /organism= Rhizobium etli CFN 42 /mol_type= genomic DNA /strain= CFN 42 /db_xref= taxon: /plasmid= p42f promoter /note= sigma54 panCp promoter; Putative transcription. initiation. gene /gene= panC /locus_tag= RHE_PF00001 CDS /EC_number= /product= pantoate beta alanine ligase protein")
18
vjimenez> more /home/genomas/pub/RE1PF/RE1PF_gene_from_GK3.dat
LocusTag GI gene_name product_name position strain gbaccession crossrefs RHE_PF GI: panC pantoate beta alanine ligase protein forward CP000138 CDD:COG0414,CDD:PF ,GI: ,InterPro:IPR003721 RHE_PF GI: panB ketopantoate hydroximethyltransferase protein forward CP CDD:COG0413,CDD:PF ,GI: ,InterPro:IPR003700 RHE_PF GI: oxyR hydrogen peroxide sensing transcriptional regulator protein, LysR family complement( ) reverse CP CDD:COG0583,CDD:PF ,CDD:PF ,GI: ,Int erPro:IPR000847,InterPro:IPR005119 RHE_PF GI: katG catalase protein forward CP CDD:COG0 376,CDD:PF ,GI: ,InterPro:IPR000763,InterPro:IPR002016
 reverse CP CDD:COG0583,CDD:PF ,CDD:PF ,GI: ,Int. erPro:IPR000847,InterPro:IPR RHE_PF00004 GI: katG catalase protein forward CP CDD:COG0. 376,CDD:PF ,GI: ,InterPro:IPR000763,InterPro:IPR")
19
use Bio::Seq::RichSeq; use Bio::SeqIO; use Bio::SeqIO::genbank;
$pathIN=$ARGV[0]; $filetoRead=$ARGV[1]; #File to Read $filetoStore=$ARGV[2]; print "Archivo $filetoRead\n"; open(OUT,">$filetoStore")|| die "Cannot open output file..$filetoStore\n"; $in = Bio::SeqIO->new(-file => "$pathIN/$filetoRead", '-format' => 'genbank'); while ((my $seq = $in->next_seq())){ $gbaccession = $seq->accession(); foreach my $f ($seq->get_SeqFeatures) { **… } close(OUT); print "# finished processing: n_of_seqs=$n_of_seqs\n"; translate_GBK_to_FileTabs4.pl
|| die Cannot open output file..$filetoStore\n ; $in = Bio::SeqIO->new(-file => $pathIN/$filetoRead , -format => genbank ); while ((my $seq = $in->next_seq())){ $gbaccession = $seq->accession(); foreach my $f ($seq->get_SeqFeatures) { **… } close(OUT); print # finished processing: n_of_seqs=$n_of_seqs\n ; translate_GBK_to_FileTabs4.pl.")
20
if($f->primary_tag() =~ /CDS/){
$posleft=$f->location->{"_start"}; $posrigth=$f->location->{"_end"}; if($f->location->{"_strand"} == 1){ $strain= "forward"; }else{ $strain= "reverse"; } if($f->has_tag('db_xref')){ $crossrefs = join(',',sort $f->each_tag_value('db_xref')); if($f->has_tag('locus_tag')){ $id = join(',',sort $f->each_tag_value('locus_tag')); if($f->has_tag('gene')){ $gene = join(',',sort $f->each_tag_value('gene')); if($f->has_tag('product')){ $product = join(',',sort $f->each_tag_value('product')); $header = $id."\t".$gi."\t".$gene."\t".$product."\t".$genepos."\t".$strain."\t".$gbaccession."\t$crossrefs\t"; print OUT "$header\n"; $n_of_seqs++;
{ $strain= forward ; }else{ $strain= reverse ; } if($f->has_tag( db_xref )){ $crossrefs = join( , ,sort $f->each_tag_value( db_xref )); if($f->has_tag( locus_tag )){ $id = join( , ,sort $f->each_tag_value( locus_tag )); if($f->has_tag( gene )){ $gene = join( , ,sort $f->each_tag_value( gene )); if($f->has_tag( product )){ $product = join( , ,sort $f->each_tag_value( product )); $header = $id. \t .$gi. \t .$gene. \t .$product. \t .$genepos. \t .$strain. \t .$gbaccession. \t$crossrefs\t ; print OUT $header\n ; $n_of_seqs++;")
21
¿y para Illumina? Generar una archivo fastq

22
Problemas con Bioperl…
La documentación de Bioperl esta incompleta Bioperl es grande (mas de 500 módulos) escrito por muchos voluntarios First, the Bioperl documentation is incomplete. In fact, until fairly recently, there was no document that provided a tutorial introduction to the project. This has changed; the bptutorial.pl document, which you’ve already seen and will see more of, is an excellent beginning, despite its occasional errors. This document cleverly combines a tutorial with quite a few example programs that you can run, as you’ll soon see. Other documentation for Bioperl is also available, including Internet-based tutorials, forthcoming books, example programs, and journal articles. So, the situation has recently improved. Second, Bioperl is big (over 500 modules), written by volunteers, and gradually evolving. The size of the project is a sign that Bioperl addresses many interesting and useful problems, but it also means that, for the new user of Bioperl, an overview of the available resources is a task in itself. The majority of the Bioperl code is quite good, especially the most-used parts of it. However, the volunteer and evolving nature of Bioperl development means that some of the code is unfinished and not as well integrated with other parts of the project as one would like. Newer or less used modules may still need some shaking out by users in real-world situations. This is where you can make an initial contribution to the project: as you find problems, report them (more on that later).
 escrito por muchos voluntarios. First, the Bioperl documentation is incomplete. In fact, until fairly recently, there was. no document that provided a tutorial introduction to the project. This has changed; the bptutorial.pl document, which you’ve already seen and will see more of, is an. excellent beginning, despite its occasional errors. This document cleverly combines a. tutorial with quite a few example programs that you can run, as you’ll soon see. Other documentation for Bioperl is also available, including Internet-based tutorials, forthcoming books, example programs, and journal articles. So, the situation has. recently improved. Second, Bioperl is big (over 500 modules), written by volunteers, and gradually. evolving. The size of the project is a sign that Bioperl addresses many interesting and. useful problems, but it also means that, for the new user of Bioperl, an overview of. the available resources is a task in itself. The majority of the Bioperl code is quite good, especially the most-used parts of it. However, the volunteer and evolving nature of Bioperl development means that. some of the code is unfinished and not as well integrated with other parts of the. project as one would like. Newer or less used modules may still need some shaking. out by users in real-world situations. This is where you can make an initial contribution. to the project: as you find problems, report them (more on that later).")
23
Referencias: Curso: Perl en Bioinformática. Autor: Bruno Contreras.
url:

Presentaciones similares


 Usted tiene – You have (Formal) El tiene – He has Ella.>")


. It features two verb changes that we will see very soon.>")
>")





 This template uses Microsofts corporate font, Segoe Segoe is not a standard font included with Windows, so if you have not.>")



>")


