Descargar la presentación
La descarga está en progreso. Por favor, espere

1
TUMOR CUTANEO SOLITARIO EN MUJER DE 30 AÑOS
M.J. López Poveda 5 de Marzo de 2.010 H.G.U. Virgen de la Arrixaca Murcia

2
Caso clínico Mujer de 30 años
Lesión cutánea nodular, eritematosa y única Localizada en MII Asintomática Pruebas complementarias sin alteraciones

12
CD68

13
S-100

14
F-XIII

16
ENFERMEDAD DE ROSAI DORFMAN CUTANEA

17
Introducción La enfermedad de Rosai–Dorfman (ERD), también conocida como histiocitosis sinusal con linfadenopatías masivas (HSLM), es una histiocitosis benigna infrecuente, caracterizada por linfofagocitosis (emperipolesis). Fue descrita por primera vez por Rosai y Dorfman en 1969 como una entidad histopatológica distinta, clínicamente caracterizada por linfadenopatías cervicales de gran tamaño, fiebre, aumento de la velocidad de eritrosedimentación, hipergammaglobulinemia policlonal y alteraciones hematológicas. Los ganglios linfáticos presentan infiltración sinusal masiva por histiocitos, con características inmunohistoquímicas típicas.
, también conocida como histiocitosis sinusal con linfadenopatías masivas (HSLM), es una histiocitosis benigna infrecuente, caracterizada por linfofagocitosis (emperipolesis). Fue descrita por primera vez por Rosai y Dorfman en 1969 como una entidad histopatológica distinta, clínicamente caracterizada por linfadenopatías cervicales de gran tamaño, fiebre, aumento de la velocidad de eritrosedimentación, hipergammaglobulinemia policlonal y alteraciones hematológicas. Los ganglios linfáticos presentan infiltración sinusal masiva por histiocitos, con características inmunohistoquímicas típicas.")
18
Epidemiología La FORMA CLÁSICA de HSLM o ERD sistémica, frecuentemente cursa con dolor y linfadenopatías bilaterales en niños o adultos jóvenes (mediana de edad: 20,6 años), con predilección en el sexo masculino (1.4:1). Se han descrito alrededor de 400 casos a nivel mundial. Más frecuente raza negra y blancos que en otras (asiáticos). Aproximadamente 23% a 45% de los pacientes presentan compromiso de, al menos, un tejido/órgano extranodal. La piel y los tejidos blandos están afectados en el 16% de los pacientes.
, con predilección en el sexo masculino (1.4:1). Se han descrito alrededor de 400 casos a nivel mundial. Más frecuente raza negra y blancos que en otras (asiáticos). Aproximadamente 23% a 45% de los pacientes presentan compromiso de, al menos, un tejido/órgano extranodal. La piel y los tejidos blandos están afectados en el 16% de los pacientes.")
19
Epidemiología La piel es la localización extraganglionar más frecuentemente afectada en ERD, pero su afectación primaria (sin linfadenopatía) es rara.
 es rara.")
20
Epidemiología de la ERDC
Entidad distinta a Enf. Rosai-Dorfman sistémica (+ jóvenes, africanos). Afectación cutánea en 11% Muy rara (pocos casos descritos): Kroumpouzos y Demierre: 27 casos de ERDC ( ): mujeres de raza blanca se afectaban con más frecuencia.Sólo dos eran asiáticos. 2002: Brenn/Calonje/Fletcher/McKee 22 pacientes: El primer estudio amplio: 22 casos de ERDC: marcada predilección por asiáticos y blancos. 2006: Frater/Maddox/Obadiah revisión 72 pacientes (+edad, mujeres, blancos y asiáticos). 2006: Wang/Chen/Liu: 21 lesiones en 6 pacientes ( ) en Taiwan: más frecuente en asiáticos. (evolución cronológica de las lesiones: -céls, -vasc, +fibrosis, + xantom.; 12 meses/12-24 meses/regresión). 2007: Yun-yi Kong/Jin-cheng Kong: 25 casos: más de 50% asiáticos.
. Afectación cutánea en 11% Muy rara (pocos casos descritos): Kroumpouzos y Demierre: 27 casos de ERDC ( ): mujeres de raza blanca se afectaban con más frecuencia.Sólo dos eran asiáticos. 2002: Brenn/Calonje/Fletcher/McKee 22 pacientes: El primer estudio amplio: 22 casos de ERDC: marcada predilección por asiáticos y blancos. 2006: Frater/Maddox/Obadiah revisión 72 pacientes (+edad, mujeres, blancos y asiáticos). 2006: Wang/Chen/Liu: 21 lesiones en 6 pacientes ( ) en Taiwan: más frecuente en asiáticos. (evolución cronológica de las lesiones: -céls, -vasc, +fibrosis, + xantom.; 12 meses/12-24 meses/regresión). 2007: Yun-yi Kong/Jin-cheng Kong: 25 casos: más de 50% asiáticos.")
21
Etiología Desconocida.
El estudio etiológico se ha centrado en la reacción histiocitaria inducida por citoquinas o infecciones por microorganismos que aún no se han identificado. Se han reportado casos de asociación con infecciones por virus herpes 6 y virus de Epstein-Barr. La hipótesis de una infección viral se correlaciona en algunos pacientes con la asociación a hipergammaglobulinema policlonal y disminución en el número de linfocitos T supresores.

22
Diagnóstico Anamnesis. Exploración física. Pruebas complementarias:
Laboratorio: ausencia de leucocitosis, anemia, hipergammaglobulinemia policlonal, aumento de la VSG, factor reumatoide positivo y niveles alterados de complemento sérico. Estudio de extensión.

23
Clínica La ERD clásica se puede presentar con una amplia variedad de manifestaciones clínicas. A pesar de ser una entidad benigna, puede comportarse de forma agresiva, generando morbimortalidad por compresión de órganos vitales. La forma de presentación más frecuente se caracteriza por linfadenopatías cervicales dolorosas de gran tamaño (90%). Suelen asociarse signos sistémicos de inflamación, como fiebre, leucocitosis, aumento de la velocidad de eritrosedimentación e hipergammaglobulinemia policlonal.
. Suelen asociarse signos sistémicos de inflamación, como fiebre, leucocitosis, aumento de la velocidad de eritrosedimentación e hipergammaglobulinemia policlonal.")
24
Clínica Cerca del 25% a 43% de los pacientes presentan compromiso extranodal. En algunos casos existe afectación extranodal sin compromiso ganglionar, que puede o no desarrollarse posteriormente con la evolución de la enfermedad. El compromiso extranodal múltiple es inusual y de mal pronóstico. La afectación extranodal más frecuente es la piel y tejidos blandos, tracto respiratorio superior, huesos y anexos paraoculares.

25
Clínica La clínica de la ERDC difiere a la clásica.
Los pacientes con ERDC cursan con buen estado general, sin fiebre, malestar, sudoración nocturna u otra sintomatología que sugiera déficit inmunitario. Lesión cutánea asintomática.

26
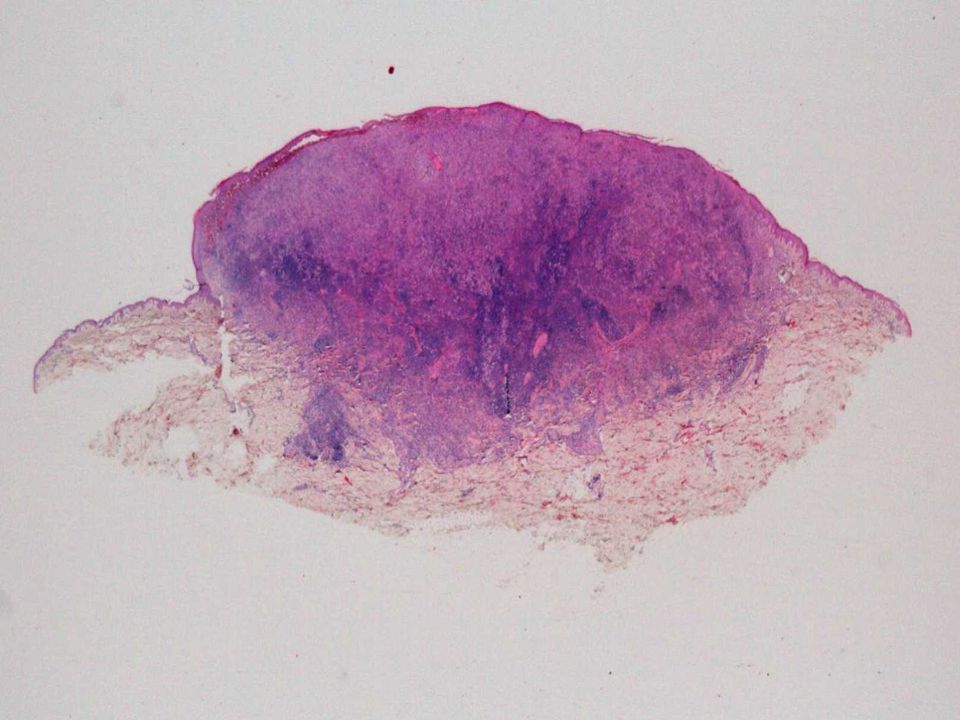
Hallazgos macroscópicos
Las manifestaciones de la ERDC tienen un amplio espectro de presentación. El espectro de lesiones histológicas varían según el tiempo de evolución. En el mismo paciente pueden ocurrir diferentes formas de presentación dependiendo de la profundidad y duración de la lesión. Es difícil distinguir la ERDC de la ERD sistémica con afectación cutánea en función de las alteraciones clínicas y/o histológicas.

27
Hallazgos macroscópicos
Pápulas o nódulos de crecimiento lento y coloración rojiza o púrpura. Placas eritematosas nodulares se forman ocasionalmente como consecuencia de la coalescencia de lesiones tras un periodo de tiempo de evolución, ocasionalmente con presencia de papulovesículas y pústulas. Placas planas hiperpigmentadas. Forma de cúpula o tumor exofítico con ulceración central.

28
Hallazgos macroscópicos. Dx diferencial clínico
Debido a esta amplia variabilidad apariencia clínica, existen múltiples patologías con las que clínicamente se puede confundir. Destacar: Linfocitoma cutis Infección fúngica Vaculitis Paniculitis Deratofibroma o dermatofibrosarcoma protuberans Sarcoidosis Tuberculosis cutis Otras histiocitosis cutáneas: incluyendo HCL, histiocitoma eruptio generalizado y el xantogranuloma.

29
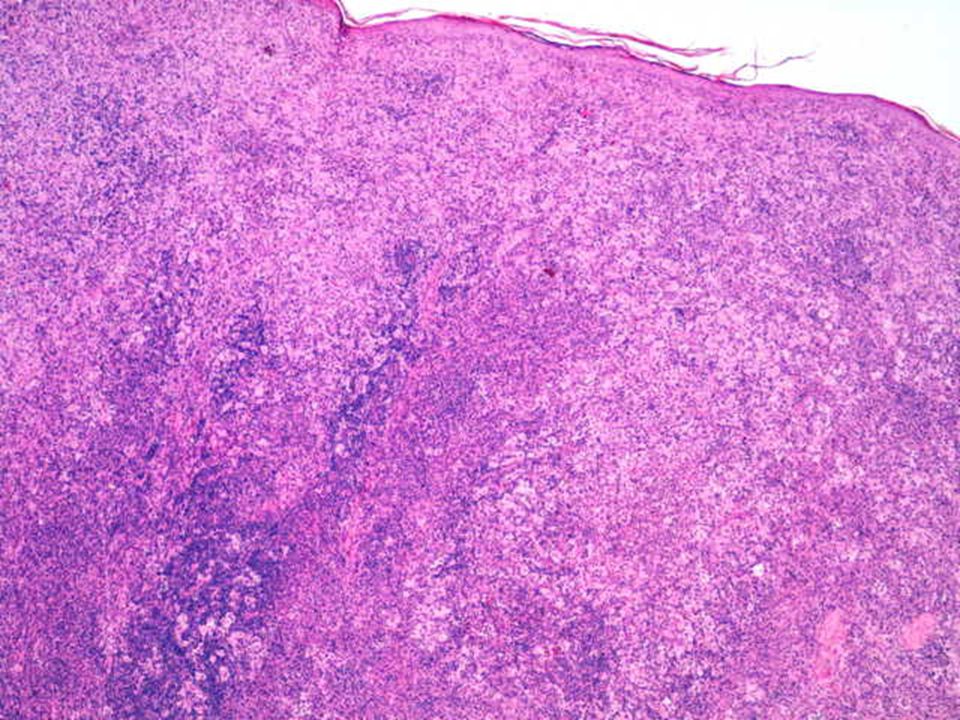
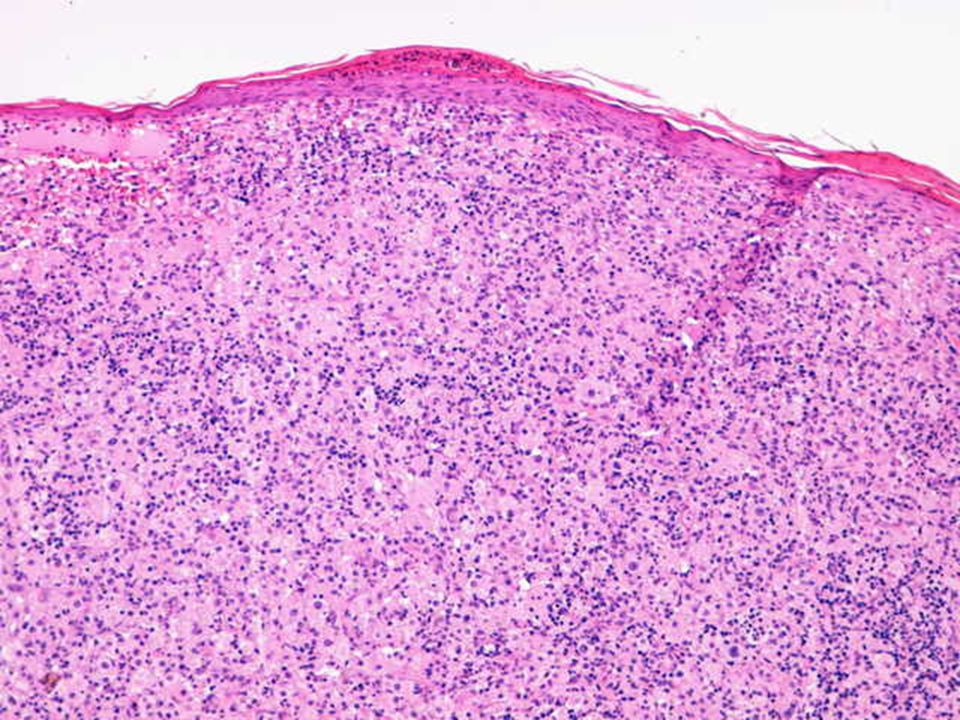
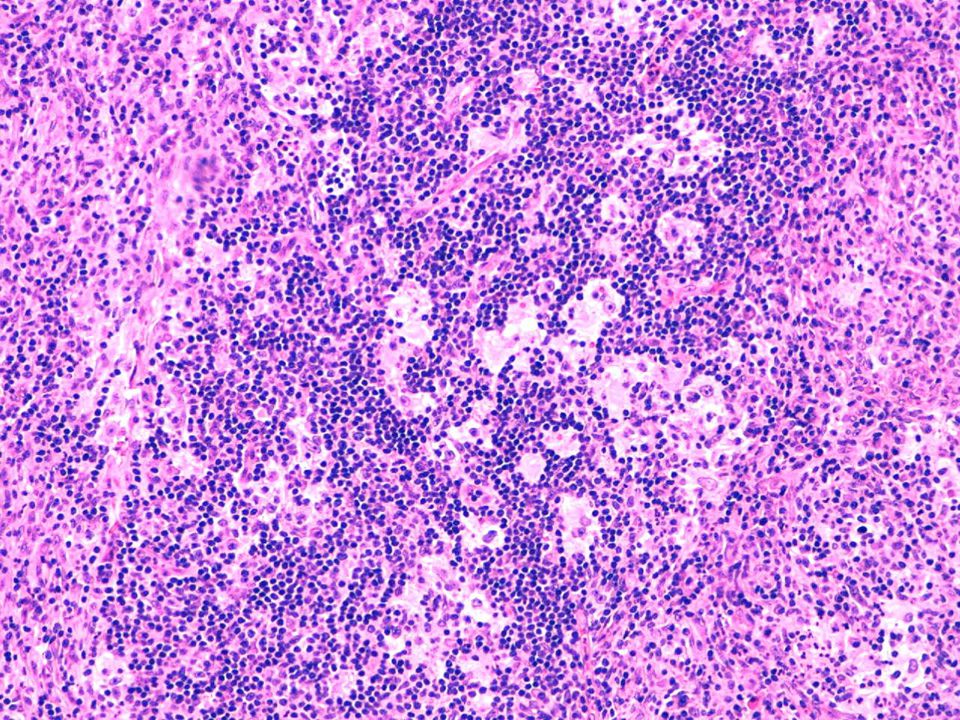
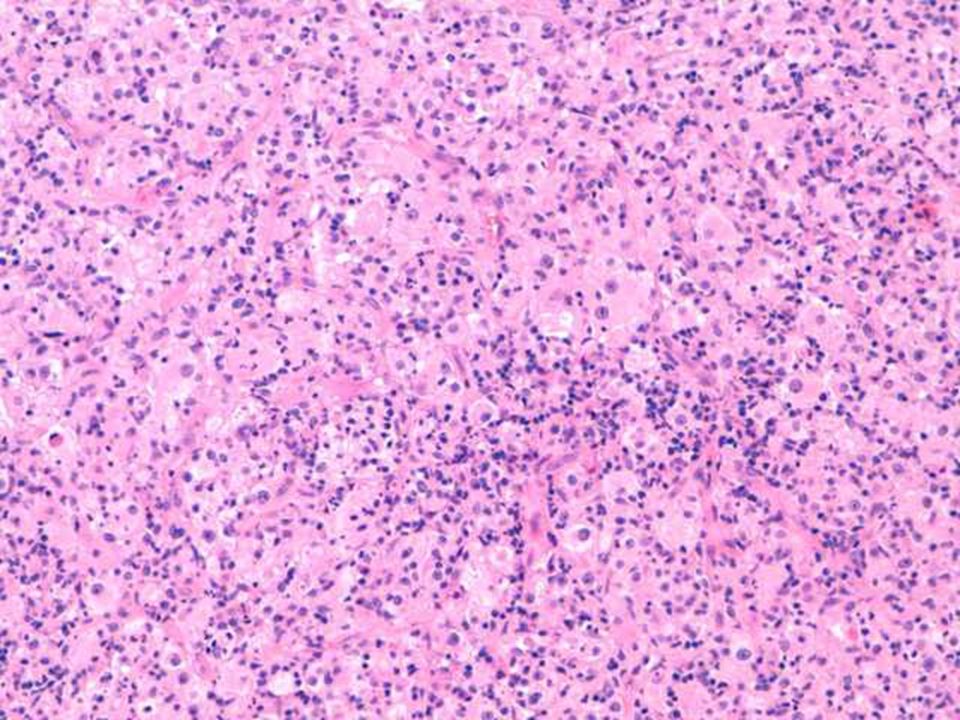
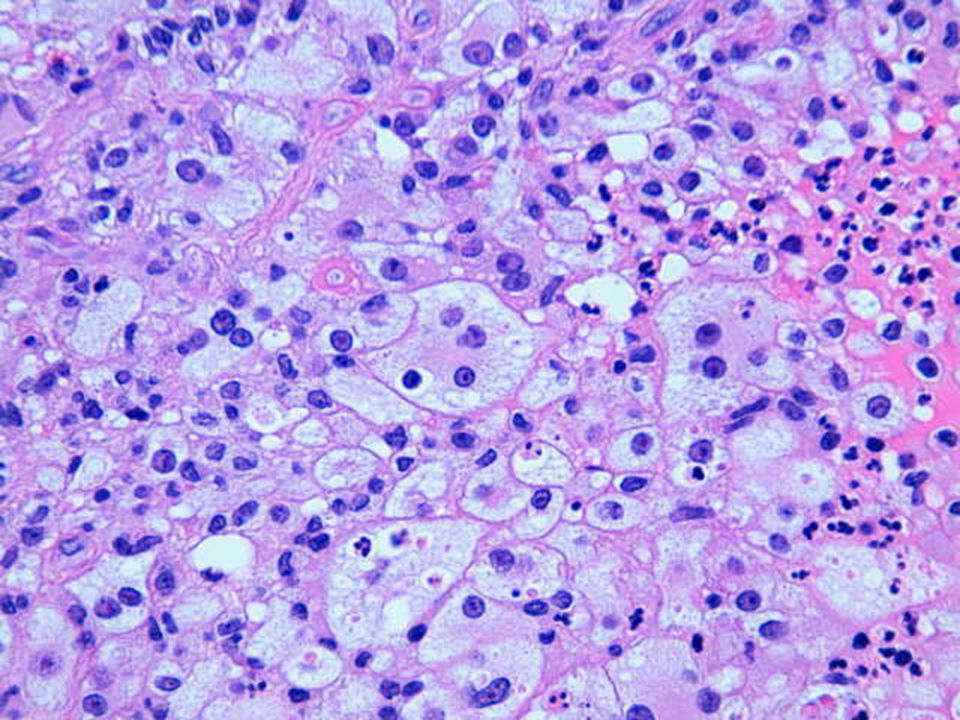
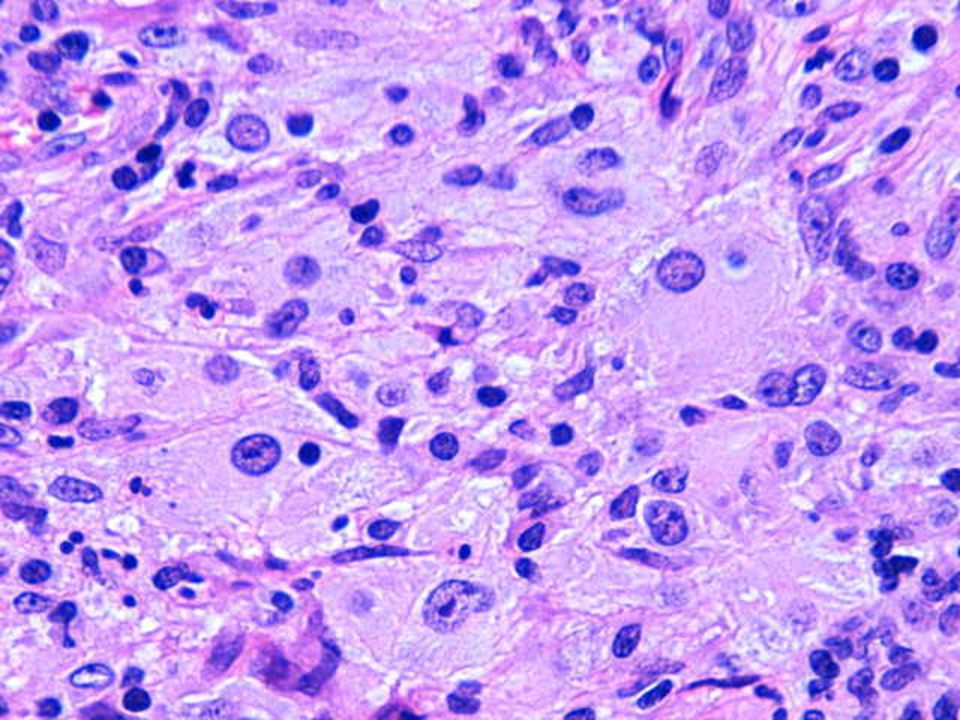
Hallazgos microscópicos
Lesiones similares tanto en ganglios como en localización extranodal. El cuadro se caracteriza por: Gran infiltrado linfohistiocitario. Histiocitos con morfología característica. Linfofagocitosis o emperipolesis. Puede existir infiltrado plasmocitario perivascular. En zonas más superficiales de la lesión puede existir un infiltrado inflamatorio mixto e inespecífico, por lo que en biopsias muy superficiales podría hacerse un diagnóstico incorrecto. Depósitos de cristales en el citoplasma de células plasmáticas y en raras ocasiones en los histiocitos. En algunas lesiones, variable grado de fibrosis.

30
Inmunohistoquímica El marcador más útil en la ERD es la proteína S100, aunque puede expresar una gran variedad de antígenos macrofágicos, tales como CD68, HAM 56, CD14, CD64 y CD15. Generalmente son negativos para CD1a.

31
Diagnóstico diferencial histológico
La enfermedad xantohistiocítica Histiocitosis maligna e histiocitosis de cels de Langerhans (con la que puede coexistir) Reticulohistiocitoma Pseudotumor inflamatorio cutáneo Lesiones fibrohistiocitoides como el histiocitoma fibroso benigno y el dermatofibrosarcoma protuberans Procesos infecciosos, inflamatorios Enfermedad de Rosai-Dorfman clásica Ver S-100, Touton, emperipolesis e infiltrado infamatorio. Ver atipia y mitosis Ver CD1a y gránulos de Birbeck Ver S100, citoplasma histiocitos e infiltrado inflamatorio Ver macroscopia y S100, emperipolesis Ver emperipolesis, plasmáticas perilesionales y S100 Clínica, exploraciones complentarias, emperipolesis, IHQ Estudio de extensión
 Reticulohistiocitoma. Pseudotumor inflamatorio cutáneo. Lesiones fibrohistiocitoides como el histiocitoma fibroso benigno y el dermatofibrosarcoma protuberans. Procesos infecciosos, inflamatorios. Enfermedad de Rosai-Dorfman clásica. Ver S-100, Touton, emperipolesis. e infiltrado infamatorio. Ver atipia y mitosis. Ver CD1a y gránulos de Birbeck. Ver S100, citoplasma histiocitos. e infiltrado inflamatorio. Ver macroscopia y S100, emperipolesis. Ver emperipolesis, plasmáticas perilesionales y S100. Clínica, exploraciones complentarias, emperipolesis, IHQ. Estudio de extensión.")
32
Evolución y tratamiento
La ERD generalmente es benigna y autolimitada, y característicamente se resuelve de forma espontánea tras varios años de evolución. No obstante, en algunos pacientes la enfermedad se presenta con alternancia de exacerbaciones y remisiones durante periodos prolongados; sin embargo, la duración de cada fase es impredecible. En un porcentaje pequeño de pacientes con ERD, en los cuales hay compromiso de órganos vitales, el tratamiento se hace necesario para detener la progresión de la enfermedad. No existe un tratamiento específico para la ERD. Esteroides, crioterapia y radioterpia. Se ha demostrado que la observación periódica es la principal intervención cuando la ERD no es masiva y no compromete órganos vitales.

33
Conclusiones La ERD es una entidad infrecuente que presenta una amplia variedad de manifestaciones clínicas. Es importante tenerla en cuenta sobre todo como diagnóstico diferencial en pacientes con múltiples linfadenopatías, habiendo descartado previamente entidades neoplásicas linfoproliferativas. La histología juega un papel importante, con características celulares clásicas y con tinciones inmunohistoquímicas que pueden orientarnos aún más para hacer el diagnóstico. Es importante destacar que la elección terapéutica se hace sobre la base de la forma de presentación y el eventual compromiso vital del paciente. Se precisan estudios más amplios para aclarar y definir la etiologia y obtener así el tto mas adecuado para esta patología.

34
GRACIAS

Presentaciones similares