Descargar la presentación
La descarga está en progreso. Por favor, espere

1
Células dendríticas

2
Células dendríticas (DC)
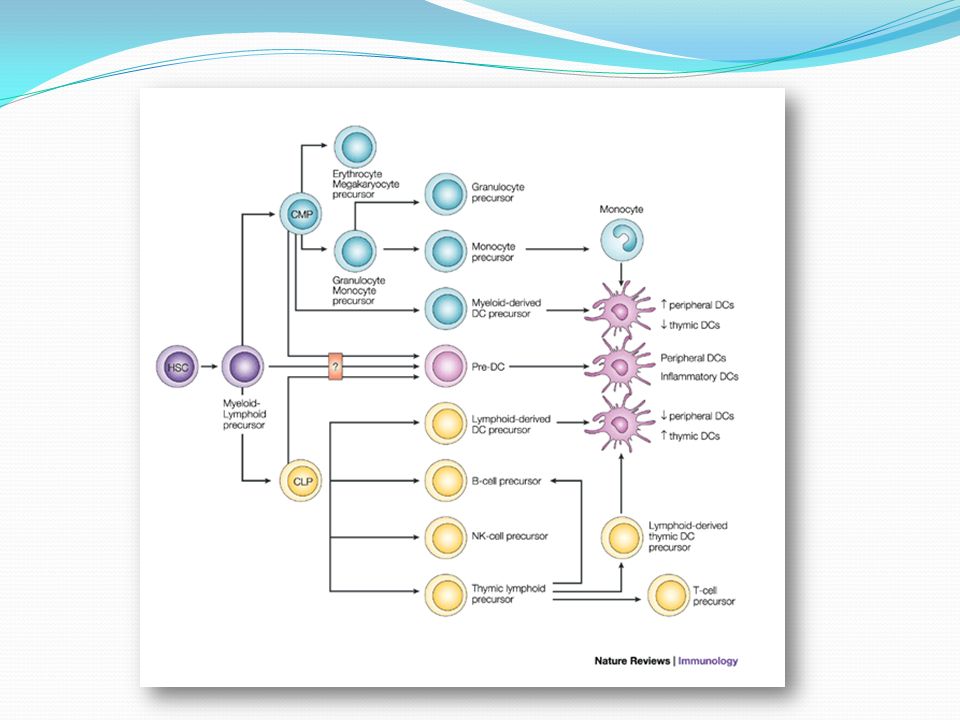
Se originan a partir de precursores hematopoyéticos CD34+. Hay 3 subtipos: Celulas dendríticas mieloides o DC1 Células dendríticas plasmocitoides o DC2 Células de Langerhans o CL Cada subtipo presenta un origen, marcadores de superficie y funciones distintas.

3
Células dendríticas (DC)
Su función principal el la presentación de Ag, la regulación de la respuesta inmune y, posiblemente, el control de los fenómenos de tolerancia inmunológica.

4
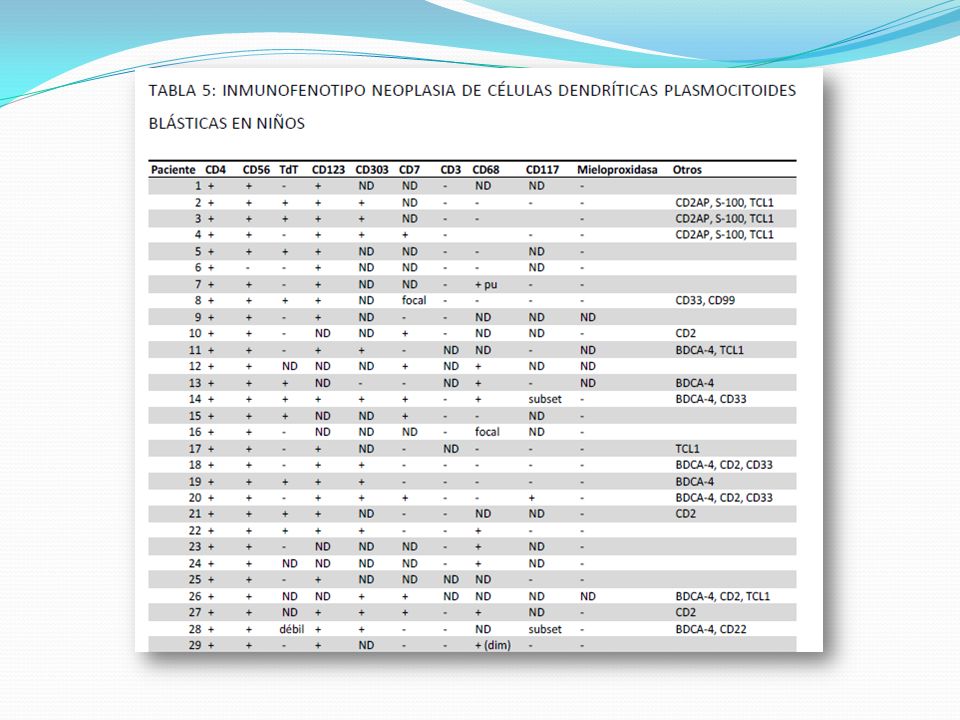
Neoplasia de células dendríticas plasmocitoide blásticas. CD4+/CD56+

5
Es una neoplasia rara que se presenta principalmente en adultos con un curso clínico desfavorable.
El cuadro inicial se manifiesta por afección cutánea y posteriormente evoluciona a una enfermedad extracutánea generalizada. Su origen se mantuvo en dilema por algunos años, y recién en el año 2001 se identificó a la célula dendrítica plasmocitoide pDC o de tipo 2 (DC2) como la célula precursora de ésta neoplasia. Actualmente se considera a esta patología como una “neoplasia hematopoyética maligna CD4+CD56+ de linaje negativo” y ha sido incluida en la clasificación de la OMS como “neoplasia de células dendríticas plasmocitoides blasticas”
 como la célula precursora de ésta neoplasia. Actualmente se considera a esta patología como una neoplasia hematopoyética maligna CD4+CD56+ de linaje negativo y ha sido incluida en la clasificación de la OMS como neoplasia de células dendríticas plasmocitoides blasticas")
6
Clasificación de la OMS
En el 2001 la OMS, en colaboración con la Sociedad de Hematopatología y la Asociación Europea de Hematopatología, publicaron una “Clasificación de Tumores de Tejidos Hematopoyéticos y Linfoides”. En el 2008, dada la rápida emergencia de nueva información biológica y genética, se ha publicado una revisión con algunas modificaciones en dicha clasificación.

7
Clasificación de la OMS de “Neoplasias mieloides y Leucemias Agudas”
Neoplasias mieloproliferativas (NMP). Neoplasias mieloides y linfoides asociadas con eosinofilia y anormalidades de PDGFRA, PDGFRB o FGFR1. Neoplasias mielodisplásicas/mieloproliferativas (SMD/NMP). Sindrome mielodisplásico (SMD). Leucemias mieloides agudas y neoplasias relacionadas. Leucemias agudas de linaje ambiguo. Leucemia/linfoma linfobláscico B. Leucemia/linfoma linfoblástico T.
. Neoplasias mieloides y linfoides asociadas con eosinofilia y anormalidades de PDGFRA, PDGFRB o FGFR1. Neoplasias mielodisplásicas/mieloproliferativas (SMD/NMP). Sindrome mielodisplásico (SMD). Leucemias mieloides agudas y neoplasias relacionadas. Leucemias agudas de linaje ambiguo. Leucemia/linfoma linfobláscico B. Leucemia/linfoma linfoblástico T.")
8
Clasificación de la OMS de “Neoplasias mieloides y Leucemias Agudas”
Neoplasias mieloproliferativas (NMP). Neoplasias mieloides y linfoides asociadas con eosinofilia y anormalidades de PDGFRA, PDGFRB o FGFR1. Neoplasias mielodisplásicas/mieloproliferativas (SMD/NMP). Sindrome mielodisplásico (SMD). Leucemias mieloides agudas y neoplasias relacionadas. Leucemias agudas de linaje ambiguo. Leucemia/linfoma linfobláscico B. Leucemia/linfoma linfoblástico T.
. Neoplasias mieloides y linfoides asociadas con eosinofilia y anormalidades de PDGFRA, PDGFRB o FGFR1. Neoplasias mielodisplásicas/mieloproliferativas (SMD/NMP). Sindrome mielodisplásico (SMD). Leucemias mieloides agudas y neoplasias relacionadas. Leucemias agudas de linaje ambiguo. Leucemia/linfoma linfobláscico B. Leucemia/linfoma linfoblástico T.")
9
Leucemias mieloides agudas y neoplasias relacionadas.
LMA con anormalidades genéticas recurrentes. LMA con cambios relacionados a mielodisplasia. Neoplasias mieloides relacionadas a terapias. LMA sin otra especificación. Sarcoma mieloide. Proliferaciones malignas relacionadas con Sindrome de Down. Blastic plasmocytoid dendritic cell neoplasma

10
Leucemias mieloides agudas y neoplasias relacionadas.
LMA con anormalidades genéticas recurrentes. LMA con t(8;21)(q22;q22), RUNX1-RUNX1T1(CBFA/ETO). LMA con inv(16)(p13q22) o t(16;16)(p13;q22), CBFB-MYH11. Leucemia promielocítica aguda con t(15;17)(q22;q11-12), PML-RARA. LMA con t(9;11)(p22;q23), MLLT3-MLL. LMA con t(6;9)(p23;q34); DEK-NUP214. LMA con inv(3)(q21q26.2) o t(3;3)(q21;q26.2), RPN1-EVI1. LMA (megacarioblástico) con t(1;22)(p13;q13), RBM15-MKL1. LMA con NPM1 mutado. LMA con CEBPA mutado. LMA con cambios relacionados a mielodisplasia. Neoplasias mieloides relacionadas a terapias. LMA sin otra especificación. LMA con diferenciación mínima. LMA sin maduración. LMA con maduración. Leucemia aguda mielomonocítica. Leucemia monoblástica y monocítica aguda. Leucemia eritroide aguda. Leucemia megacarioblástica aguda. Leucemia basofílica aguda. Panmielosis aguda con mielofibrosis. Sarcoma mieloide. Proliferaciones malignas relacionadas con Sindrome de Down. Blastic plasmocytoid dendritic cell neoplasma
(q22;q22), RUNX1-RUNX1T1(CBFA/ETO). LMA con inv(16)(p13q22) o t(16;16)(p13;q22), CBFB-MYH11. Leucemia promielocítica aguda con t(15;17)(q22;q11-12), PML-RARA. LMA con t(9;11)(p22;q23), MLLT3-MLL. LMA con t(6;9)(p23;q34); DEK-NUP214. LMA con inv(3)(q21q26.2) o t(3;3)(q21;q26.2), RPN1-EVI1. LMA (megacarioblástico) con t(1;22)(p13;q13), RBM15-MKL1. LMA con NPM1 mutado. LMA con CEBPA mutado. LMA con cambios relacionados a mielodisplasia. Neoplasias mieloides relacionadas a terapias. LMA sin otra especificación. LMA con diferenciación mínima. LMA sin maduración. LMA con maduración. Leucemia aguda mielomonocítica. Leucemia monoblástica y monocítica aguda. Leucemia eritroide aguda. Leucemia megacarioblástica aguda. Leucemia basofílica aguda. Panmielosis aguda con mielofibrosis. Sarcoma mieloide. Proliferaciones malignas relacionadas con Sindrome de Down. Blastic plasmocytoid dendritic cell neoplasma.")
11
Neoplasias a células dendríticas plasmocitoides blásticas
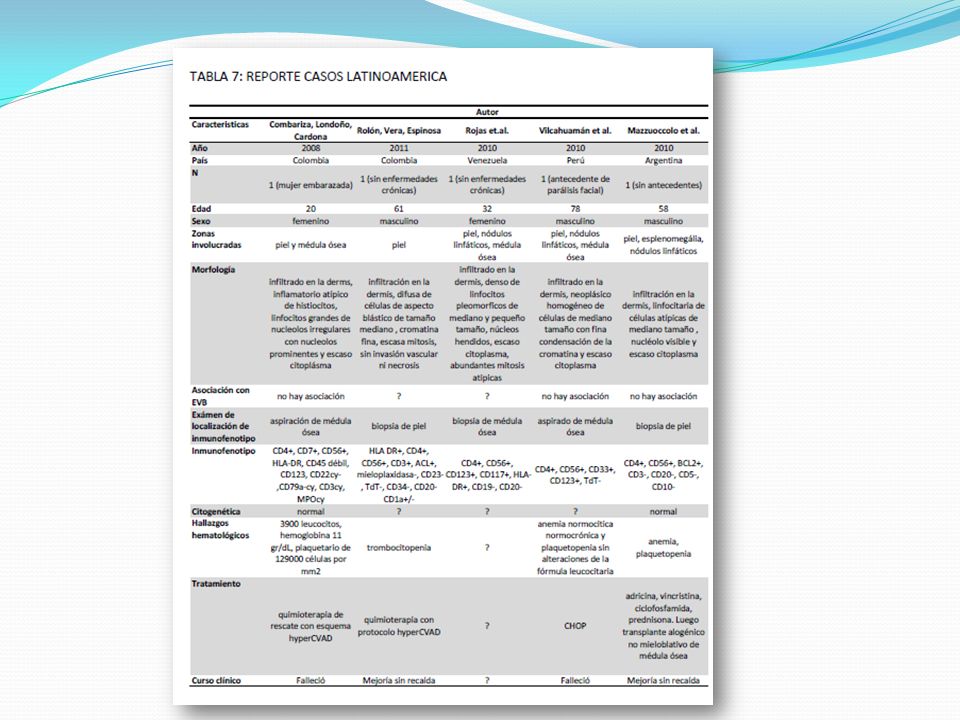
En el 2002 una serie de 23 leucemias, con un inusual pero único inmunofenotipo fue reportado por el “Grupo de Estudio Inmunológicos de Leucemias Frances” (GEIL). Las células malignas fueron positivas para CD4 y CD56, pero ningunos pertenecían al linaje de linfocitos T, ni al linaje mielomonocítico. Estos casos tenían muchas características clínicas y biológicas comunes: La mayoría de los pacientes eran personas mayores. Se presentaban con lesiones cutáneas, compromiso de la MO y frecuente evolución hacia una leucemia manifiesta. El curso clínico fue siempre agresivo, conduciendo a la muerte dentro de los 3 años, a pesar de la respuesta inicial al tratamiento. Las células malignas aparecían morfológicamente inmaduras, con un citoplasma agranular, lleno de microvesículas. El análisis histológico reveló que éstas células infiltran la dermis pero no la epidermis, y en los nódulos linfáticos, se localizan en áreas interfoliculares. Fueron frecuentes las anomalías citogenéticas. No se observó asociación con infección con EBV.
. Las células malignas fueron positivas para CD4 y CD56, pero ningunos pertenecían al linaje de linfocitos T, ni al linaje mielomonocítico. Estos casos tenían muchas características clínicas y biológicas comunes: La mayoría de los pacientes eran personas mayores. Se presentaban con lesiones cutáneas, compromiso de la MO y frecuente evolución hacia una leucemia manifiesta. El curso clínico fue siempre agresivo, conduciendo a la muerte dentro de los 3 años, a pesar de la respuesta inicial al tratamiento. Las células malignas aparecían morfológicamente inmaduras, con un citoplasma agranular, lleno de microvesículas. El análisis histológico reveló que éstas células infiltran la dermis pero no la epidermis, y en los nódulos linfáticos, se localizan en áreas interfoliculares. Fueron frecuentes las anomalías citogenéticas. No se observó asociación con infección con EBV.")
13
ANTECEDENTES Desde 1989 hasta el 2002 un total de 18 artículos han informado de 58 casos de enfermedades malignas que muestran características similares y perfil fenotípico celular característico: CD4+CD56+CD3-CD13-CD33-CD19-. Se le han asignados diferentes nombres, entre ellos: Linfoma histiocítico o neoplasia hematológica asociada a histicitos. Linfoma cutáneo agranular CD4+CD56+. Neoplasia hematodérmica agranular CD4+CD56+. Leucemia/linfoma de células NK blásticas. Linfoma NK Linfoma relacionado a células precursoras mielomonocítica. Se propuso como célula de origen a un precursor de células NK, un precursor mielomonocítico inmaduro o un precursor NK/mielomonocítico mixto. Esto se basaban únicamente en la positividad exclusiva de uno o dos marcadores asociados a linaje (CD56 para NK, CD68 y CD36 para monocitos) pero no a un inmunofenotipo completo consistente con un equivalente definido normal. Así la OMS en el 2001 consideró esta entidad como “Linfoma de células NK blásticas”. Luego se identificó el homólogo normal de esas células malignas como células dendríticas plasmocitoides (pDC) y en la revisión del 2008 de la OMS ya aparecen como “Neoplasia de células dendríticas plasmocitoides blásticas”.
 pero no a un inmunofenotipo completo consistente con un equivalente definido normal. Así la OMS en el 2001 consideró esta entidad como Linfoma de células NK blásticas . Luego se identificó el homólogo normal de esas células malignas como células dendríticas plasmocitoides (pDC) y en la revisión del 2008 de la OMS ya aparecen como Neoplasia de células dendríticas plasmocitoides blásticas .")
15
Manifestaciones Clínicas
Se presenta en adultos mayores, pero la enfermedad puede desarrollarse en todas las edades. En un estudio reciente 67% adultos > 50 años 7% adultos entre 36 y 50 años 18% en jóvenes entre 18 y 35 años Además se reportaron casos en niños (4) y lactantes(3). Afecta más a hombres que a mujeres (3:1) Curso clínico agresivo y desfavorable, con un tiempo de sobrevida no mayor a 3 años.
 y lactantes(3). Afecta más a hombres que a mujeres (3:1) Curso clínico agresivo y desfavorable, con un tiempo de sobrevida no mayor a 3 años.")
16
Manifestaciones Clínicas
Inicialmente puede caracterizarse por afección cutánea con presencia de nódulos único o multiples, de unos pocos milimetros a 10 cm y aspecto variable (placa, pápulas, moretón o nódulos subcutáneos) Las lesiones nodulares suelen ser eritematosas, hiperpigmentadas, enrojecidas o violáceas, presentar descamación fina y en algunas ocaciones ulceradas.
 Las lesiones nodulares suelen ser eritematosas, hiperpigmentadas, enrojecidas o violáceas, presentar descamación fina y en algunas ocaciones ulceradas.")
17
Manifestaciones Clínicas
La mayoría de los casos evolucionan a la enfermedad extracutánea generalizada, que se caracteriza por la aparición de linfoadenopatías, hepato y/o esplenomegalia e infiltración de MO, pero cualquier órgano o tejido puede finalmente encontrarse afectado La lesión típica es cutánea. El compromiso de la MO es sumamente frecuente ya sea en la presentacion o durante el curso de la enfermedad. Los organos linfoides extranodales tambien son frecuentemente invadidos: bazo, hígado, amígdalas y tejidos asociados a mucosas (nasofaríngeo, encías, conjuntiva ocular o mucosa bronquial). Además, mas raramente puede invadir otros como SCN, corazón, pulmón, riñón, etc.
. Además, mas raramente puede invadir otros como SCN, corazón, pulmón, riñón, etc.")
19
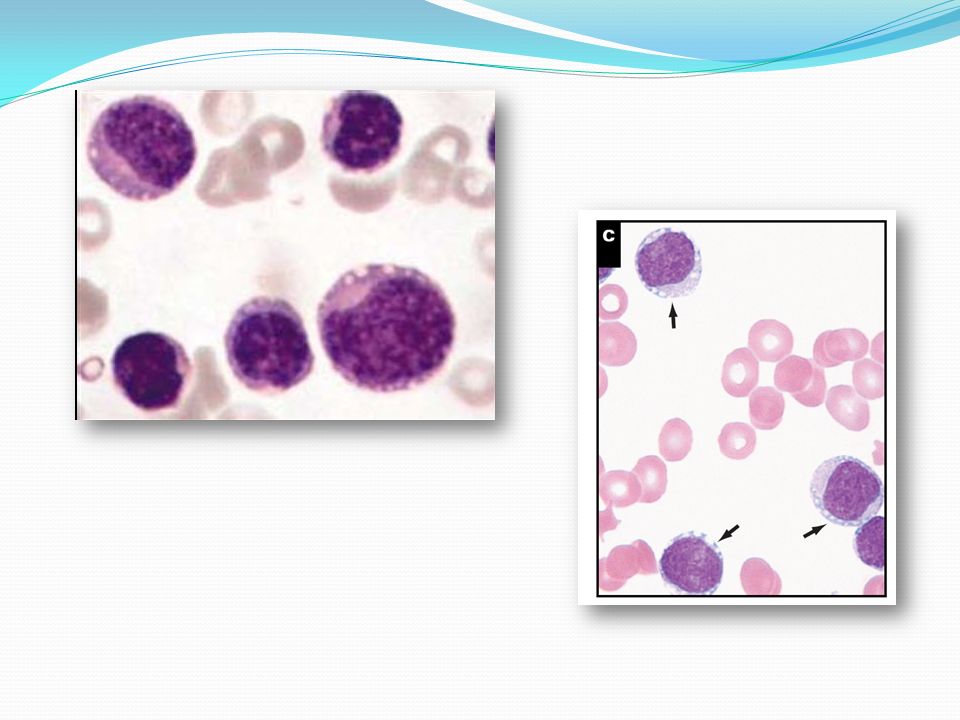
Características microscópicas
La lesión cutánea se caracteriza por la presencia de células neoplásicas que infiltran toda la dermis hasta el tejido celular subcutáneo sin afectar la capa epidérmica. Las células neoplásicas presentan núcleo de tamaño mediano, redondo u oval, con cromatina laxa y en ocasiones nucleolos pequeños por lo que pueden confundirse con linfo o mieloblastos. El citoplasma suele ser basófilo y no muy abundante. En frotis sanguíneo las células suelen observarse con núcleo excéntrico y microvacuolas.

21
Hallazgos hematológicos
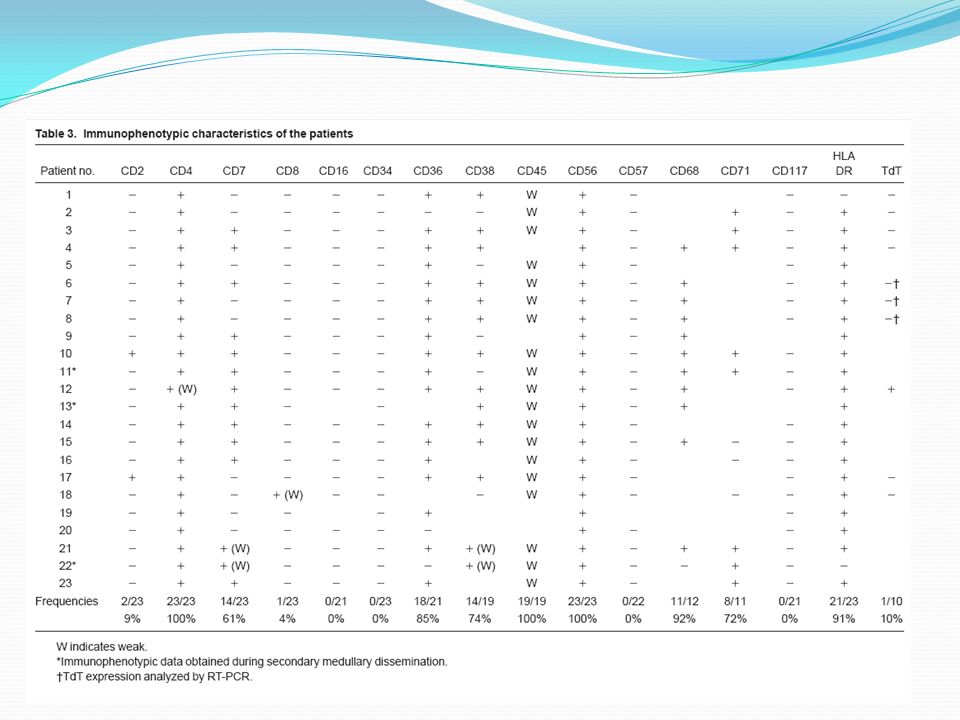
En la serie de Feuillard: El 91% mostró citopenia. El 78% presentó trobocitopenia. El 34% ánemia. El 34% neutropenia.

22
Citogenética Las alteraciones genéticas observadas son inespecíficas ya que se han observado en otras neoplasias y pueden abarcar regiones muy grandes del genoma. Se han identificado diversas anomalías cromosómicas e las pDC malignas como: Cariotipos anormales (44XY, 45XY) 13q+ Deleciones 5q y de los cromosomas 13 y15. Mas recientemente se identificaron deleciones de 6q,12p,13q, 15q y cromosoma 9, asi como deleciones y ganancias en región 12p13.
 13q+ Deleciones 5q y de los cromosomas 13 y15. Mas recientemente se identificaron deleciones de 6q,12p,13q, 15q y cromosoma 9, asi como deleciones y ganancias en región 12p13.")
24
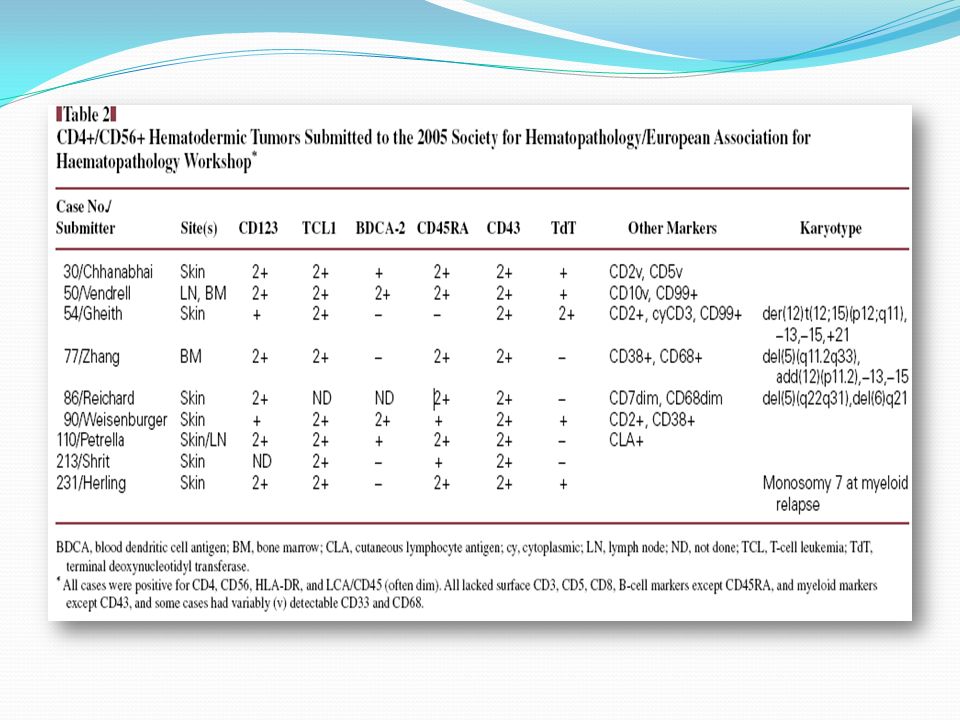
Inmunofenotipo Para determinar el origen de las células malignas se seleccionaron pacientes en la base de un fenotipo homogéneo. Por ej: CD4+CD56+CD3-CD5-CD19-CD13-CD33- Es un perfil inmunofenotípico único y no se corresponde con ninguna conocida T, B, NK o subconjunto mieloide en cualquier etapa de diferenciación. Por el contrario, coincide exactamente con la de precursores de pDC presentes en sangre u organos linfoides

27
Comparación de Inmunofenotipo de células malignas y pDC normal.
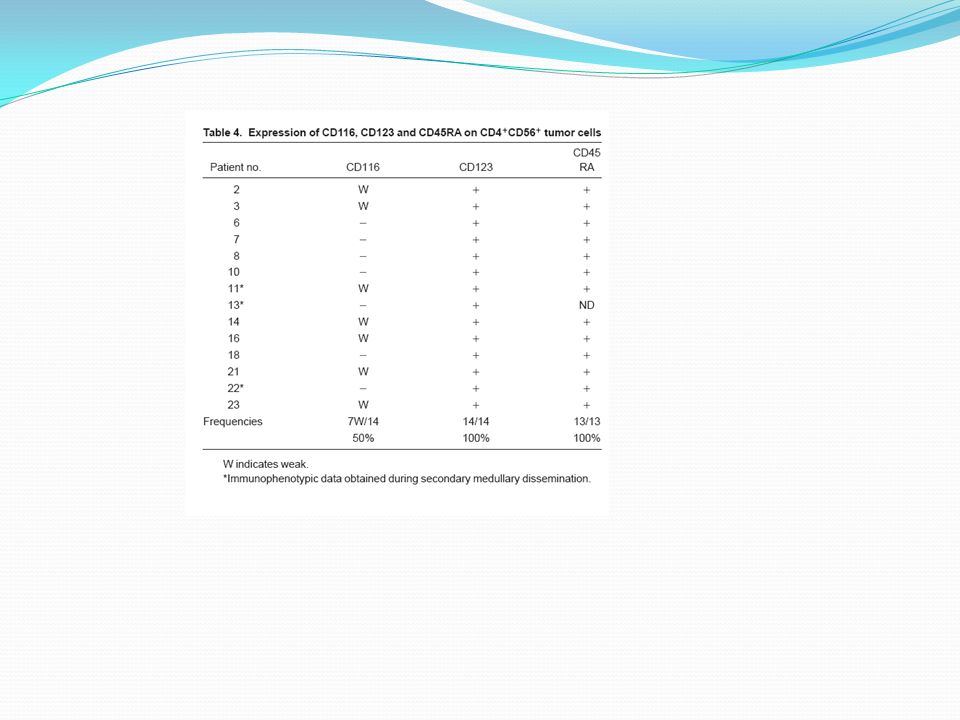
Specific markers for Normal pDC Malignant pDC T or NK lynphocytes CD2 subset + or - CD3 - CD5 CD7 CD4 + CD8 CD56 CD16 CD57 CD94 TIA1 Granzyme-B Perforine Myeloid lineage CD13 CD33 Low CD11c MPO CD14 CD64 CD36 CD68 Specific markers for Normal pDC Malignant pDC B lynphocytes CD19 - CD20 CD22 CD79a Ig mb or cy pDC HLA-DR + CD45RA CD45RO CD116(GM-CSF Ra) Low CD123(IL-3Ra) BDCA-2 + or - BDCA-4 Immature/mature DC CD32 CD80 CD86 CD40 CD1a CD1c CD83 Comparación de Inmunofenotipo de células malignas y pDC normal. La presencia de CD56 fue la primer diferencia importante. Al igual que las pDC normales expresan HLA-DR y otras moléculas asociadas a la presentación de Ag (CD40, CD86). Está presente CD32 (marcador involucrado en captura de Ag vía Ac Ig G). Exhiben marcadores característicos de pDC normales elevadamente positivos (CD123, CD45RA, BDCA).
 Low. CD123(IL-3Ra) BDCA-2. + or - BDCA-4. Immature/mature DC. CD32. CD80. CD86. CD40. CD1a. CD1c. CD83. Comparación de Inmunofenotipo de células malignas y pDC normal. La presencia de CD56 fue la primer diferencia importante. Al igual que las pDC normales expresan HLA-DR y otras moléculas asociadas a la presentación de Ag (CD40, CD86). Está presente CD32 (marcador involucrado en captura de Ag vía Ac Ig G). Exhiben marcadores característicos de pDC normales elevadamente positivos (CD123, CD45RA, BDCA).")
28
Otras similitudes de las células malignas con las pDC normales
Expresan mRNA similares. Presentan igual comportamiento invitro. En cultivo con IL-3 y CD40-L, o con virus adquieren morfología típica de DC con múltiples dendritas finas alrededor de toda la célula. Se ponen positivas CD1a, CD1c and CD83 y las moléculas involucradas en la co-estimulación de linfocito T o presentación de Ag son inducidas o altamente reguladas. La estimulación de las células también demostró dos propiedades fundamentales que inequívocamente la ligan al linaje pDC. En respuesta al virus de la Influenza son capaces de sintetizar IFN-α En un estado mas diferenciado (después de una estimulación) son capaces de activar linfocitos naive de sangre de cordón
 son capaces de activar linfocitos naive de sangre de cordón.")
29
Asociación de células tumorales al linaje pDC basado en criterios inmunofenotípicos
Las observaciones del grupo GEIL fueron fundadas en mostrar marcadores característicos de pDC: HLA-DR + CD123 high CD45RA + CDD5RO – BDCA-2 + BDCA-4 + Pero se han reportados muchos otros casos con perfiles similares e iguales características citológicas, histológicas y clínicas, con ligeras diferencias inmunofenotípicas. Estas diferencias principalmente consideran la expresión de: CD33 + (9 casos) CD3cy + (11 casos) CD94 + (2 casos) TIA1 + (4 casos) CD56 – (2 casos)
 CD3cy + (11 casos) CD94 + (2 casos) TIA1 + (4 casos) CD56 – (2 casos)")
31
CD33: diferencia DC mieloide de linfoide.
Recientemente se confirmó que es un Ag que puede estar débilmente expresado en pDC de sangre periférica normal. Por lo tanto, “la expresión de CD33 no excluye el diagnóstico de pDC maligno y podría no ser considerado como un marcador aberrante”. CD3cy: es considerado como un marcador específico del linaje de linfocitos T. El linfocito pre-T expresa CD7 en asociación con CD3cy, luego CD2 y finalmente CD5. Los CD4 y CD8 se adquieren después. 7 de 11 pacientes fueron CD7+, solo 1 de 8 fueron CD2+ mientras que los 9 casos testeados fueron CD5-. Éntonces se descarta un origen a células T (además el inmunofenotipo CD4+CD56+ no es clásico en el linaje T) CD94 y TIA1: marcador de NK. Su presencia en asociación con CD56 plantea nuevamente un origen de linaje NK, pero no expresan otros marcadores de NK (CD16, CD57, perforinas) y el citoplasma siempre está libre de gránulos azurófilos. Entonces podría pensarse que el CD56 no es un marcador específico de los NK. Además la presencia de CD4 no es compatible con un origen NK. CD56: es un marcador asociado a NK. Se ha documentado muy bien un caso de pDC maligno con sólo 10% de CD56 y otro con fenotipo CD4+CD56-lin-
 CD94 y TIA1: marcador de NK. Su presencia en asociación con CD56 plantea nuevamente un origen de linaje NK, pero no expresan otros marcadores de NK (CD16, CD57, perforinas) y el citoplasma siempre está libre de gránulos azurófilos. Entonces podría pensarse que el CD56 no es un marcador específico de los NK. Además la presencia de CD4 no es compatible con un origen NK. CD56: es un marcador asociado a NK. Se ha documentado muy bien un caso de pDC maligno con sólo 10% de CD56 y otro con fenotipo CD4+CD56-lin-")
32
Blom y colab han propuesto una vía de desarrollo para pre-pDC normales a partir de stem cells CD34+ CD4+ y describieron 4 estadíos principales. CD34 CD45RA CD123 Progenitor temprano ++ - + Progenitor tardío Pro-pDC Pre-pDC

34
Blom y colab han propuesto una vía de desarrollo para pre-pDC normales a partir de stem cells CD34+ CD4+ y describieron 4 estadíos principales. CD34 CD45RA CD123 Progenitor temprano ++ - + Progenitor tardío Pro-pDC Pre-pDC Exhiben la morfología de Células plasmáticas y secretan gran cantidad de IFN-α Se observó que las células malignas CD4+CD56+lin- tienen una baja expresión de CD45 en todos los casos al igual que otras neoplasias malignas hematopoyéticas, y son positiva para CD34 o TdT en varios casos. La morfología de las DC malignas se parece a otros blastos mieloides o linfoides y no a células plasmáticas. De acuerdo a lo anterior podría proponerse que las células pDC malignas son detenidas en su maduración muy cerca del estadío de pro-pDC Las pDC CD4+CD56+lin- son detenidas en un estadío temprano de maduración.

35
Respuesta al tratamiento y pronóstico
La mayoría de los casos presenta una buena respuesta inicial a la quimioterapia (80-90%) pero sufren una o varias recaídas en pocos meses (media de 9-11meses) que evolucionan rápidamente hacia la muerte (< 3 años). Raramente han sido reportados remisiones de tiempo mas prolongado o curas esporádicas. Generalmente en pacientes jóvenes que han recibido terapia de inducción para leucemias agudas seguida de transplante de stem cell alogénica en la primera remisión completa.
 pero sufren una o varias recaídas en pocos meses (media de 9-11meses) que evolucionan rápidamente hacia la muerte (< 3 años). Raramente han sido reportados remisiones de tiempo mas prolongado o curas esporádicas. Generalmente en pacientes jóvenes que han recibido terapia de inducción para leucemias agudas seguida de transplante de stem cell alogénica en la primera remisión completa.")
36
Conclusiones Las neoplasias CD4+CD56+lin- son raros. Los hallazgos inmunofenotípicos, morfológicos, citogenéticas y clínicos revelaron muchas características comunes correspondiente a un nueva entidad, de curso agresivo y corta supervivencia. El diagnóstico puede sospecharse a partir de dichas características, pero ninguna es especifica por si sola. El diagnostico final requiere un inmunofenotipo compatible. Las pDC en un estadío temprano de diferenciación fue identificado como la contraparte normal de las células tumorales. El origen y patogénesis de esta neoplasia apenas está empezando a esclarecerse, pero aún quedan muchas incógnitas por resolver respecto a la función de las DC normales y su contraparte neoplásica.

40
MUCHAS GRACIAS

Presentaciones similares



















, bcl2 s Ig, CD19,CD20,CD21 SOBREVIDA 7-9 AÑOS TRASFORMACION.>")




