Descargar la presentación
La descarga está en progreso. Por favor, espere

1
UNIVERSIDAD NACIONAL DE ASUNCIÓN FACULTAD DE CIENCIAS QUÍMICAS
CÁTEDRA DE INMUNOLOGÍA CLÍNICA UNIDAD TEMÁTICA: INMUNODEFICIENCIAS

2
OBJETIVOS Definir y reconocer las características generales de las inmunodeficiencias Reconocer la relación entre el tipo de infecciones y el compromiso inmunitario subyacente en un paciente inmunodeprimido Comprender la racionalidad de los estudios inmunológicos solicitados para la caracterización de un paciente inmunodeficiente Comprender la etiopatogenia del SIDA y las diferentes fases de la infección por el HIV Conocer ciertos aspectos básicos relativos a las complejas interacciones que se establecen entre el HIV y el sistema inmunitario del paciente infectado

3
CONTENIDOS Generalidades. Clasificación Inmunodeficiencias primarias
Inmunodeficiencias de predominio humoral Inmunodeficiencias combinadas celulares Inmunodeficiencias de función fagocitaria Inmunodeficiencias del complemento Inmunodeficiencias asociadas a otro defecto Generalidades de tratamiento

4
Niño con infecciones recurrentes
Causas : Retraso en maduración fisiológica Falta de lactancia materna Cambios a ambientes contaminados Enfermedades alérgicas Defectos estructura/función Inmunodeficiencias primarias

5
Inmunodeficiencia del Lactante
Es temporal y su recuperación depende de : Estímulos no patógenos del medio ambiente Lactancia materna Experiencia inmunológica materna Programación inmunológica Se puede retrasar : Uso exagerado de antimicrobianos Asepsia estricta Falta de lactancia materna

6
Inmunodeficiencias Primarias
David Vetter ( )
")
7
Datos de Alarma Infecciones frecuentes : > 8 otitis / año
> 2 sinusitis / año > 2 neumonías / año Abscesos recurrentes > 2 meses con uso AB Necesidad AB intravenosos Detención de peso y talla Ulceras orales recurrentes Onfalorrexis > 1 mes

8
Datos de Alarma Otitis media recurrente Otitis perforada
Supuración crónica Sinusitis recurrente Gérmenes no habituales Evolución tórpida

9
Datos de Alarma Uso crónico de AB (2 meses o más) con poca mejoría
Neumonías recurrentes Gérmenes atípicos o habituales pero de evolución no habitual

10
Datos de Alarma Retraso en crecimiento (peso y talla) y desarrollo
Abscesos cutáneos recurrentes o en órganos profundos

11
Datos de Alarma Requiere AB intravenosos para poder mejorar
Ulceras orales o cutáneas recurrentes después del 1er año Requiere AB intravenosos para poder mejorar

12
Datos de Alarma Infecciones persistentes Onfalorrexis tardía
Antecedentes familiares de casos previos de IDP

13
Inmunodeficiencias secundarias.
Déficit nutricional Stress: neurohormonas, corticoides Exo / endotoxinas Virus (HIV, Sarampión, Varicela, EBV) Pérdida de barreras de defensa : Continuidad de piel Inflamación en mucosas
 Pérdida de barreras de defensa : Continuidad de piel. Inflamación en mucosas.")
14
Inmunodeficiencias secundarias.
Medicamentos : Inmunosupresión, neutropenia Mutilación : Esplenectomía, timectomía Padecimientos células inmunes : Leucemia, linfoma, Enf. autoinmunes

15
Mitos y Realidades ¿ Son raras o pasan desapercibidas ?
Deficiencia IgA = 1 / 500 hab > 100 diferentes IDP En conjunto : (Pediatría) Frec. Comparable a Leucemia Frec. Comparable a Linfomas 4 veces > frecuente que Fibr. quística WHO Sci Group Report. 1992;3:195
 Frec. Comparable a Leucemia. Frec. Comparable a Linfomas. 4 veces > frecuente que Fibr. quística. WHO Sci Group Report. 1992;3:195.")
16
Diagnóstico Temprano Evita daño a órganos vitales :
Infección Brutton Bronquiectasias Autoinmunidad Ataxia-Telangiectasia Neumonitis Hiper IgM Anemia hemolítica

17
Bronquiectasias

18
Infecciones en 138 pacientes con IDP
Carneiro Sampaio, M. Interasma XVI Buenos Aires. Octubre 1999.

19
Grupos de IDP Predominio humoral Combinadas (celulares) Fagocitarias
Complemento Asociadas

20
IDP de predominio humoral

21
Inmunodeficiencias de predominio humoral
Agammaglobulinemia congénita Enfermedad de Brutton Defecto en la maduración de linfocitos B IgG, IgM, IgA, muy bajas o ausentes Herencia ligada al X Infecciones después de 6 m

22
Agammaglobulinemia ligada al X
Defecto en el gen btk Tirosin quinasa de la Enf. de Brutton Falla en la maduración linfocitos B Disminución de CD19

23
Inmunodeficiencias de predominio humoral
Hipogammaglobulinemia con hiper IgM Defecto en CD40 ligando (CD154) Defecto en CD40 o vías de activación IgG e IgA bajas IgM normal o elevada Infecciones recurrentes
 Defecto en CD40 o vías de activación. IgG e IgA bajas. IgM normal o elevada. Infecciones recurrentes.")
24
Hipogammaglobulinemia con Hiper IgM
CD40 ligando CD154 Familia TNFR

25
Hipogammaglobulinemia con Hiper IgM

26
Hipogammaglobulinemia con Hiper IgM
Dx temprano Tx temprano Px vida normal

27
Inmunodeficiencias de predominio humoral
Deficiencia selectiva de IgA Deleción del gen para cadenas pesadas alfa Puede ser asintomática o asociarse a alergia Deficiencia de subclases de IgG Cuantitativa (IgG1, IgG2, IgG3, IgG4) Funcional hacia polisacáridos
 Funcional hacia polisacáridos.")
28
Asma en pacientes con IDP
203 niños con asma moderada - grave 4 niños con Deficiencia de IgA Frecuencia 1:50 (10 veces mayor) 3 pacientes con otras IDP : Def IgA + Def IgG2 Inmunodeficiencia común variable Deficiencia de antipolisacáridos Excelente respuesta IGIV Carneiro Sampaio, M. Interasma XVI Buenos Aires. Octubre 1999.
 3 pacientes con otras IDP : Def IgA + Def IgG2. Inmunodeficiencia común variable. Deficiencia de antipolisacáridos. Excelente respuesta IGIV. Carneiro Sampaio, M. Interasma XVI Buenos Aires. Octubre")
29
Def. de síntesis de antipolisacáridos con Ig normales
Susceptibilidad anormal a infecciones Otitis media Niveles séricos normales IgA, IgM, IgG, subclases de IgG Incremento postvacunal < 4X basales Vacuna 23 serotipos Str pneumoniae Respuesta 4 a 6 sem después Respuesta % serotipos

30
Inmunodeficiencias humorales
Estudios Iniciales : Ig séricas totales IgG IgM IgA IgE Isohemaglutininas (IgM antiA /antiB)
")
31
Inmunodeficiencias humorales
Estudios avanzados : Subclases de IgG Respuesta específica a vacunación polisacáridos (Neumococo, H.influenzae) proteínas (tétanos, difteria, polio) CD40 ligando (CD154), CD19
 proteínas (tétanos, difteria, polio) CD40 ligando (CD154), CD19.")
32
Def. de síntesis de antipolisacáridos con Ig normales
10 niños con S. de deficiencia IgG2 específica Carneiro Sampaio, M. Interasma XVI Buenos Aires. Octubre 1999.

33
Respuesta de antipolisacáridos a 6 serotipos
Masculino de 4 años de edad Otitis media recurrente 2 Neumonías en el último año Falta de respuesta a Ag polisacáridos de neumococo Carneiro Sampaio, M. Interasma XVI Buenos Aires. Octubre 1999.

34
IDP combinadas (celulares)
")
35
Inmunodeficiencias Combinadas
Disgenesia reticular La más grave de todas Defecto maduración Aplasia medular en recién nacido

36
Inmunodeficiencias Combinadas
Inmunodeficiencia combinada severa IDCS Ligada al X Defecto en la cadena gamma del receptor común de interleucinas (IL-2, IL-4, IL-7, IL-9, IL-15) Deficiencia de ADA (adenosin desaminasa) Defecto en el metabolismo de bases púricas (DNA)
 Deficiencia de ADA (adenosin desaminasa) Defecto en el metabolismo de bases púricas (DNA)")
37
Cadena común gamma

38
IDCS

39
Deficiencia de ADA

40
Inmunodeficiencias celulares.
Estudios iniciales : Cuenta de linfocitos (BH, rosetas T) PC de hipersensibilidad tardía (PPD, Candidina, Histoplasmina, Coccidiodina)
 PC de hipersensibilidad tardía (PPD, Candidina, Histoplasmina, Coccidiodina)")
41
Inmunodeficiencias celulares.
Estudios avanzados : Subpoblaciones de linfocitos CD3, CD4, CD8 Marcadores de activación CD25 HLA clase I y clase II Transformación de linfocitos con mitógenos (PHA) o con Ag (PPD, tétanos, Candida)
 o con Ag (PPD, tétanos, Candida)")
42
IDP de función fagocitaria

43
Inmunodeficiencias Fagocitarias
Neutropenia congénita Continua Cíclica Enfermedad granulomatosa crónica Defecto en enzimas de la bomba oxidativa Abscesos recurrentes Hepatoesplenomegalia

44
EGC Defecto Cit b558 gp91 phox p22 phox p47 phox p67 phox

45
Abscesos recurrentes Granulomas

47
Inmunodeficiencias Fagocitarias
Deficiencia de adhesinas Defecto en CD18 Cadena beta 2 de integrinas LFA1 (CD11a / CD18) Mac1 (CD11b / CD18) P150/95 (CD11c / CD18) Leucocitosis persistente Onfalorrexis tardía (> 1 mes)
 Mac1 (CD11b / CD18) P150/95 (CD11c / CD18) Leucocitosis persistente. Onfalorrexis tardía (> 1 mes)")
48
Onfalitis (onfalorrexis tardía)
")
49
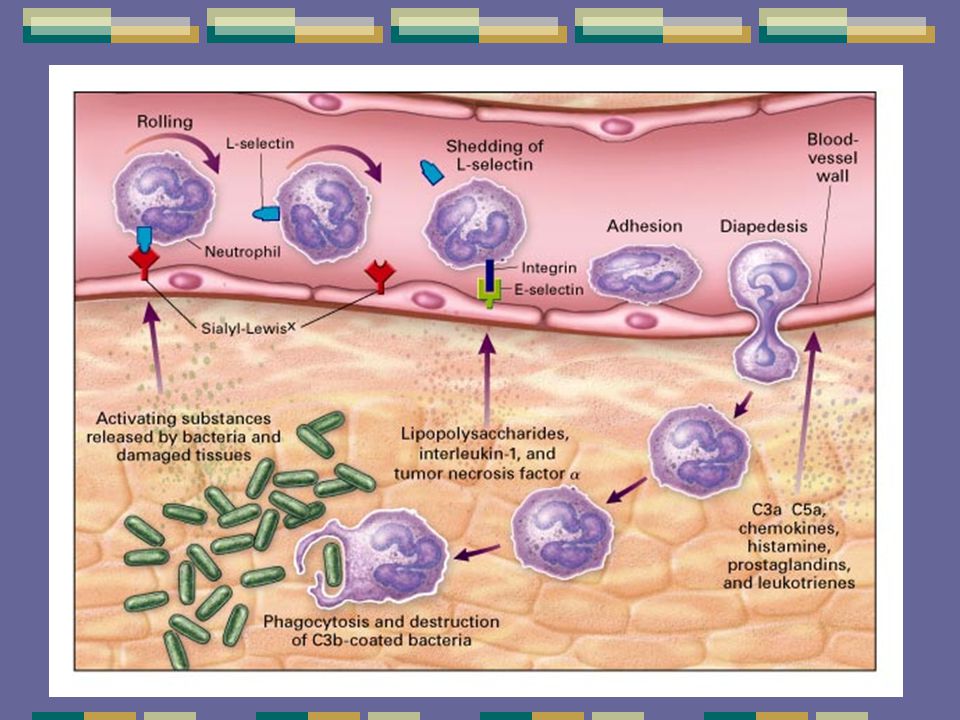
Bacterias ¿Leucocitos?

50
Inmunodeficiencias fagocitarias
Estudios iniciales : Cuenta de neutrófilos ( seriada ) Morfología de neutrófilos Reducción NBT (nitroazul de tetrazolio)
 Morfología de neutrófilos. Reducción NBT (nitroazul de tetrazolio)")
51
Reducción de NBT negativa

52
Reducción positiva (Formazán)
")
53
Inmunodeficiencias fagocitarias
Estudios avanzados : CD18 (deficiencia de adhesinas) Fagocitosis de levaduras Quimiotaxis in vitro Quimioluminiscencia, flujocitometría (Aniones de bomba oxidativa, DHR)
 Fagocitosis de levaduras. Quimiotaxis in vitro. Quimioluminiscencia, flujocitometría (Aniones de bomba oxidativa, DHR)")
54
Dehidrorodamina (DHR)
")
55
Fagocitosis de levaduras
Quimiotaxis de leucocitos

56
IDP del Complemento

57
Deficiencias del Complemento
Deficiencia de C2 Autoinmunidad (pobre depuración de CIC) Deficiencia de inhibidor de C1q Angioedema hereditario (aumento C3a y C5a) Deficiencia de C9 Infecciones por Neisserias (deficiencia MAC)
 Deficiencia de inhibidor de C1q. Angioedema hereditario (aumento C3a y C5a) Deficiencia de C9. Infecciones por Neisserias (deficiencia MAC)")
58
Deficiencias del Complemento

59
Deficiencias de Complemento
Estudios iniciales : CH50 C3 C4 50%

60
Fijación de complemento
Hemólisis

61
Deficiencias de Complemento
Estudios avanzados : Determinación de componentes Pruebas de inhibidor de C1q Cuantitativas / Cualitativas

62
IDP Asociadas a otro defecto

63
Inmunodeficiencias Asociadas
Ataxia telangiectasia Ataxia progresiva irreversible Telangiectasias oculares, cutáneas Infecciones respiratorias recurrentes Defecto en linfocitos T Deficiencia de IgA, IgG2 Radiosensibilidad susceptibilidad a CANCER Herencia autosómica recesiva (ATM)
")
64
Telangiectasias oculares

65
Ataxia Telangiectasia
Herencia autosómica recesiva Riesgo en c / embarazo : 50% Portadores 25% Afectados Gen ATM ¿ CA de mama ?

66
Inmunodeficiencias Asociadas
S. de Wiskott Aldrich Herencia ligada al X Eczema Trombocitopenia Plaquetas pequeñas Infecciones respiratorias recurrentes Defecto en linfocitos T, elevación de IgE

67
S. de Wiskott Aldrich WASP Proteína del S. de W-A

68
Inmunodeficiencias Asociadas
S. de DiGeorge Hipoplasia o aplasia de timo Deficiencia de linfocitos T Ausencia paratiroides (PTH) Hipocalcemia neonatal Crisis convulsivas de difícil control Cardiopatía congénita leve o grave
 Hipocalcemia neonatal. Crisis convulsivas de difícil control. Cardiopatía congénita leve o grave.")
69
Bolsas y Arcos faríngeos
Cara Paratiroides Timo Aorta A. pulmonar Ventrículos

70
S. De DiGeorge

71
Tetralogía de Fallot

72
S. de DiGeorge

73
Mitos y Realidades ¿ No hay nada que hacer ?
Buena calidad de vida con uso constante de GGIV en IDP humorales Sobrevida 80% en IDP mixtas graves con TMO antes de los 4 meses de edad Consejo Genético

74
Inmunodeficiencias primarias
Generalidades de tratamiento : Aislamiento Evitar vacunas de virus vivos o BCG Antimicrobianos profilácticos Productos radiados (3000 rads)
")
76
Inmunodeficiencias Primarias
Manejo de pacientes con I.D.P. Uso de gammaglobulina intravenosa Transplante de médula ósea Terapia génica

77
Transplante de médula ósea
HLA idéntico (hermano gemelo) HLA haploidéntico (padre/madre) HLA compatible (Depletado de linfocitos T) Transplante de células totipotenciales (Sangre de cordón umbilical)
 HLA haploidéntico (padre/madre) HLA compatible. (Depletado de linfocitos T) Transplante de células totipotenciales (Sangre de cordón umbilical)")
78
Transplante de M.O.

79
Transplante de M.O.

80
Transplante de M.O.

81
Transplante de células totipotenciales
Fuentes de obtención: Médula ósea Sangre periférica Cordón umbilical Hígado fetal (< 13 semanas) Postnatal / Prenatal
 Postnatal / Prenatal.")
82
Transplante prenatal (in útero)
1988 : 1er. Transplante Antecedentes familiares IDP Dx prenatal de linfocito desnudo Ausencia de HLA en linfocitos Infusión 7 ml de 16 x 106 células Vena umbilical (guiado por U/S) Presencia de linfocitos con HLA-A9 Touraine JL. Lancet 1989; 1: 1382
 Presencia de linfocitos con HLA-A9. Touraine JL. Lancet 1989; 1:")
83
Administración intravenosa
Uso de CSF Aféresis x 10 9 leucos Separar células CD34+ Transfección Gen normal Administración intravenosa

84
Métodos para introducir DNA
Ruptura física de la membrana Transporte con liposomas Vectores virales recombinantes: Virus de papiloma simiano (SV40) Adenovirus, retrovirus, lentivirus... Shangara L. Immunology Today 2000; 7: 180-9
 Adenovirus, retrovirus, lentivirus... Shangara L. Immunology Today 2000; 7:")
85
Terapia Génica

86
Deficiencia de ADA Ashanti da Silva 1990 1er. Paciente Tx Edad 13 años

87
Enfermedad Granulomatosa Crónica
Forma de Herencia : Autosómica Ligada al X Terapia Génica

88
Consejo Genético Permite conocer riesgo de recurrencia Requisitos :
Dx temprano Dx preciso (molecular) Asesoría por Médico Genetista
 Asesoría por Médico Genetista.")
89
Autosómica Dominante

90
Autosómica Recesiva

91
Ligada al X

93
Bibliografía Inmunología fundamentos. Roitt Iván. Editorial Panamericana 9 edición. 2002 Inmunología. Goldsby, R; Richard, A; Kindt, Tomas J; Osbome, Barbara. 4 edición Mac Gaw Hill 2002. Introducción a la inmunología humana. Fainboim, Geffner. Editorial panamericana sexta edición 2011

Presentaciones similares







 ESPECÍFICAS (Respuesta inmunitaria) – La unión antígeno anticuerpo es específica.>")













