Descargar la presentación
La descarga está en progreso. Por favor, espere

2
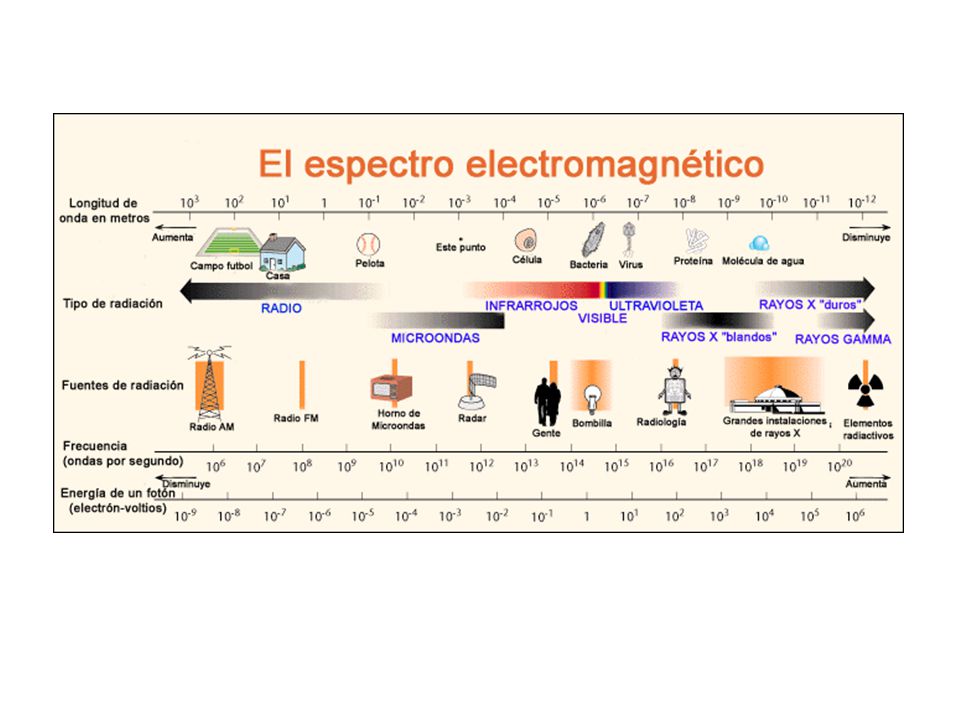
Los rayos X Se suministran unos 50 kV como diferencia de potencial (alto voltaje) entre un filamento incandescente (por el que se hace pasar una corriente i de bajo voltaje, unos 5 A a unos 12 V) y un metal puro (normalmente Cu o Mo), estableciéndose entre ambos una corriente de unos 30 mA de electrones libres. Desde el filamento incandescente (cargado negativamente) saltan electrones hacia el ánodo (cargado positivamente) provocando, en los átomos de este último, una reorganización electrónica en sus niveles de energía. Tubos convencionales de rayos X que se utilizan en los laboratorios de Cristalografía
 entre un filamento incandescente (por el que se hace pasar una corriente i de bajo voltaje, unos 5 A a unos 12 V) y un metal puro (normalmente Cu o Mo), estableciéndose entre ambos una corriente de unos 30 mA de electrones libres. Desde el filamento incandescente (cargado negativamente) saltan electrones hacia el ánodo (cargado positivamente) provocando, en los átomos de este último, una reorganización electrónica en sus niveles de energía. Tubos convencionales de rayos X que se utilizan en los laboratorios de Cristalografía.")
3
El restablecimiento energético del electrón anódico que se excitó, se lleva a cabo con emisión de rayos X con una frecuencia que corresponde exactamente al salto de energía concreto (cuántico) que necesita ese electrón para volver a su estado inicial. Estos rayos X tienen por tanto una longitud de onda concreta y se conocen con el nombre de radiación característica. Las radiaciones características más importantes en Cristalografía de rayos X son las llamadas líneas K-alpha (Kα), donde los electrones caen a la capa más interior del átomo (mayor energía de ligadura). Sin embargo, además de estas longitudes de onda concretas, se produce también todo un espectro de longitudes de onda, muy próximas entre sí, y que se denomina radiación contínua, debido al frenado por el material de los electrones incidentes. Distribución de longitudes de onda de los rayos X que se producen en tubos convencionales de rayos X en donde el material anódico es cobre (Cu), molibdeno (Mo), cromo (Cr) ó wolframio (W) . Sobre el llamado espectro contínuo aparecen las llamadas líneas características K-alpha (Kα) y K-beta (Kβ). El comienzo de los espectros contínuos aparece a una longitud de onda que es aproximadamente 12.4/V, en donde V representa la diferencia de potencial (en kV) entre filamento y ánodo. Para una misma tensión entre ánodo y filamento, sólo se excitan las radiaciones características del molibdeno (figura de la izquierda).
, molibdeno (Mo), cromo (Cr) ó wolframio (W) . Sobre el llamado espectro contínuo aparecen las llamadas líneas características K-alpha (Kα) y K-beta (Kβ). El comienzo de los espectros contínuos aparece a una longitud de onda que es aproximadamente 12.4/V, en donde V representa la diferencia de potencial (en kV) entre filamento y ánodo. Para una misma tensión entre ánodo y filamento, sólo se excitan las radiaciones características del molibdeno (figura de la izquierda).")
4
Simetría de los cristales
La simetría es la constancia, la repetición de algo en el espacio y/o en el tiempo, como en las guardas, pétalos de una flor, la sucesión de noche y dia, una pieza musical, etc. Concretándonos a los objetos finitos, existen varias operaciones (elementos de simetría) que describen las repeticiones. En las guardas nos encontramos con operaciones de traslación (el motivo se repite por traslación). La repetición de los pétalos de las flores nos conduce a operaciones de giro (el motivo se repite por giro) alrededor de ejes de simetría y, aunque no exactamente, la simetría que nos muestra una nos llevaría a considerar las operaciones denominadas planos de simetría (la operación que ocurre cuando uno se mira en un espejo). Análogamente, por ejemplo, si nos fijamos en la relación entre los objetos tridimensionales de la figura de abajo, descubriremos un nuevo elemento de simetría denominado centro de simetría, que sería el punto imaginario colocado entre ambos objetos: Dos objetos relacionados por un centro de simetría
 que describen las repeticiones. En las guardas nos encontramos con operaciones de traslación (el motivo se repite por traslación). La repetición de los pétalos de las flores nos conduce a operaciones de giro (el motivo se repite por giro) alrededor de ejes de simetría y, aunque no exactamente, la simetría que nos muestra una nos llevaría a considerar las operaciones denominadas planos de simetría (la operación que ocurre cuando uno se mira en un espejo). Análogamente, por ejemplo, si nos fijamos en la relación entre los objetos tridimensionales de la figura de abajo, descubriremos un nuevo elemento de simetría denominado centro de simetría, que sería el punto imaginario colocado entre ambos objetos: Dos objetos relacionados por un centro de simetría.")
5
Combinando estos elementos de simetría con las traslaciones características de un cristal (que veremos más adelante), surgen nuevos elementos de simetría con componentes de deslizamiento (ejes helicoidales y planos de deslizamiento). Poliedro mostrando un eje de rotación binario que pasa por los centros de las aristas de arriba y abajo Poliedro mostrando un plano de simetría que relaciona la parte de arriba con la de abajo

6
Representación gráfica de las 32 clases cristalinas
Las clases centrosimétricas aparecen con la palabra Laue

7
Redes directa y recíproca
Cualquier distribución repetitiva de un objeto o motivo, viene caracterizada por el conjunto de las traslaciones que lo repiten periódicamente. A este conjunto de traslaciones lo denominamos red directa. Las traslaciones que describen las repeticiones en los cristales pueden expresarse como una combinación lineal de tres traslaciones básicas, no coplanares, es decir, independientes, que denominamos ejes reticulares. Estos ejes definen un paralelogramo (en 2 dimensiones), o un paralelepípedo (en 3 dimensiones) que se denomina celda unidad. Este área elemental (en el caso de 2 dimensiones), o volumen elemental (en el caso de 3 dimensiones), que encierra la parte mínima de la distribución, genera, mediante traslaciones, la distribución completa, que en el caso que nos ocupa (3 dimensiones) se llama cristal. Celda elemental definida por las 3 traslaciones no coplanares denominadas ejes reticulares
, o un paralelepípedo (en 3 dimensiones) que se denomina celda unidad. Este área elemental (en el caso de 2 dimensiones), o volumen elemental (en el caso de 3 dimensiones), que encierra la parte mínima de la distribución, genera, mediante traslaciones, la distribución completa, que en el caso que nos ocupa (3 dimensiones) se llama cristal. Celda elemental definida por las 3 traslaciones no coplanares denominadas ejes reticulares.")
8
Dentro de la celdilla, y debido a los elementos de simetría de la distribución, hay una parte mínima (unidad asimétrica) que, por aplicación de la simetría, genera la celdilla unidad. Un motivo estructural o unidad asimétrica El motivo estructural de la izquierda se repite mediante los elementos de simetría, en este caso un eje helicoidal La repetición del motivo (unidad asimétrica) genera el contenido de la celdilla elemental La repetición de celdillas elementales genera la totalidad del cristal
 genera el contenido de la celdilla elemental. La repetición de celdillas elementales genera la totalidad del cristal.")
9
Familia de planos reticulares que cortan al eje vertical de la celdilla en 2 partes y al eje horizontal en 1 parte. Estos planos son paralelos al tercer eje reticular que no se muestra en la figura. Familia de planos reticulares que cortan al eje vertical de la celdilla en 3 partes y al eje horizontal en 1 parte. Estos planos son paralelos al tercer eje reticular que no se muestra en la figura. El número de partes en que una familia de planos corta a los ejes de la celdilla puede asociarse con un triplete de números que identifica a la familia de planos. En las figuras anteriores, los cortes, y por tanto los tripletes, serían (210) y (310), según los ejes vertical, horizontal y perpendicular a la figura. En esta figura, los índices de los planos dibujados serían (022), es decir, que esa familia de planos no corta al eje a, y corta a los ejes b y c en 2 partes iguales, respectivamente.
 y (310), según los ejes vertical, horizontal y perpendicular a la figura. En esta figura, los índices de los planos dibujados serían (022), es decir, que esa familia de planos no corta al eje a, y corta a los ejes b y c en 2 partes iguales, respectivamente.")
10
RED RECÍPROCA Cualquier plano puede caracterizarse, también, por un vector (σhkl) perpendicular a él. Por lo tanto, la proyección del vector de posición de cualquier punto del plano sobre esta perpendicular es constante e independiente del punto; es la distancia al origen de ese plano, es decir, su espaciado (dhkl ). De todos los vectores proporcionales, que son normales a un plano, si tomamos (como σhkl) el de módulo 1/dhkl, σhkl representa a toda la familia de planos hkl de interespaciado dhkl, de forma que se cumple el producto: |σhkl| dhkl = 1. Si definimos que el módulo del vector σhkl es 1/dhkl, el producto de ese vector, por el espaciado dhkl de la familia de planos, es la unidad. Si tomamos un vector, 2 veces más largo que σhkl , el espaciado de la familia de planos que representa, será la mitad.
 perpendicular a él. Por lo tanto, la proyección del vector de posición de cualquier punto del plano sobre esta perpendicular es constante e independiente del punto; es la distancia al origen de ese plano, es decir, su espaciado (dhkl ). De todos los vectores proporcionales, que son normales a un plano, si tomamos (como σhkl) el de módulo 1/dhkl, σhkl representa a toda la familia de planos hkl de interespaciado dhkl, de forma que se cumple el producto: |σhkl| dhkl = 1. Si definimos que el módulo del vector σhkl es 1/dhkl, el producto de ese vector, por el espaciado dhkl de la familia de planos, es la unidad. Si tomamos un vector, 2 veces más largo que σhkl , el espaciado de la familia de planos que representa, será la mitad.")
11
A partir de este vector normal, de módulo 1/dhkl, si tomamos otro que sea un número entero (n) de veces más largo, para mantener que el producto del módulo de σhkl por dhkl sea la unidad, éste nuevo vector (n.σhkl ) corresponderá a un espaciado n veces menor que el primero y por lo tanto describiría a la familia de planos nh,nk,nl. De esta manera, resulta que los vectores normales (σhkl) son recíprocos a los espaciados interplanares. Los extremos de estos vectores forman también una red periódica de puntos, que por esa propiedad de reciprocidad se llama red recíproca de la red original de traslaciones. Los puntos recíprocos así obtenidos reciben el triplete de números hkl (índices de Miller) que representa a la correspondiente familia de planos. Generación de algunos puntos recíprocos de una red. Por claridad del dibujo el tercer eje de la red directa (c) sería perpendicular al dibujo. Las líneas rojas representan a los planos cuyos índices se indican en azul. Por ejemplo, el punto recíproco de índices (3,1,0) está situado sobre el vector perpendicular al plano (3,1,0) y su distancia al origen O es inversamente proporcional al espaciado de dicha familia de planos. De este modo, la red directa y sus planos están solidariamente asociados con la red recíproca. Además, sobre esta red recíproca se puede definir también una celda (celda recíproca) cuyas traslaciones periódicas vienen determinadas por tres ejes recíprocos que forman entre sí unos ángulos recíprocos. ¿Por qué este nuevo concepto: la red recíproca? Porque representar una familia de planos por un sólo punto ya es algo que parece simplificar las cosas; y otra razón importante es que nos servirá para obtener un modelo geométrico, muy sencillo, que interpreta el fenómeno de la difracción en los cristales.
 son recíprocos a los espaciados interplanares. Los extremos de estos vectores forman también una red periódica de puntos, que por esa propiedad de reciprocidad se llama red recíproca de la red original de traslaciones. Los puntos recíprocos así obtenidos reciben el triplete de números hkl (índices de Miller) que representa a la correspondiente familia de planos. Generación de algunos puntos recíprocos de una red. Por claridad del dibujo el tercer eje de la red directa (c) sería perpendicular al dibujo. Las líneas rojas representan a los planos cuyos índices se indican en azul. Por ejemplo, el punto recíproco de índices (3,1,0) está situado sobre el vector perpendicular al plano (3,1,0) y su distancia al origen O es inversamente proporcional al espaciado de dicha familia de planos. De este modo, la red directa y sus planos están solidariamente asociados con la red recíproca. Además, sobre esta red recíproca se puede definir también una celda (celda recíproca) cuyas traslaciones periódicas vienen determinadas por tres ejes recíprocos que forman entre sí unos ángulos recíprocos. ¿Por qué este nuevo concepto: la red recíproca Porque representar una familia de planos por un sólo punto ya es algo que parece simplificar las cosas; y otra razón importante es que nos servirá para obtener un modelo geométrico, muy sencillo, que interpreta el fenómeno de la difracción en los cristales.")
12
Dispersión y difracción
La difracción (de los rayos X) es el fenómeno físico a través del cual se manifiesta la interacción fundamental de los rayos X con los cristales (materia ordenada). Dispersión por un átomo Llamamos factor atómico de dispersión (factor de "scattering") a la razón entre la amplitud dispersada por un átomo y la de un electrón aislado. Como la velocidad de los electrones en el átomo es mucho mayor que la variación del vector eléctrico de la onda, la radiación sólo "ve" una nube electrónica media, que viene caracterizada por la densidad electrónica de carga ρ(r). Si esta distribución se considera esféricamente simétrica, sólo depende de la distancia al núcleo Dispersión por una red monoatómica: difracción Cuando el agregado de átomos está estructurado según una red periódica tridimensional, de forma que los átomos constituyen nudos de esta red, las relaciones geométricas precisas entre los átomos del agregado dan lugar a diferencias de fase muy particulares. Se producen composiciones cooperativas entre las ondas dispersadas y la muestra actúa como una red de difracción de tres dimensiones. En estas condiciones, los efectos de interferencia externa producen efectos de dispersión estructurados en picos de intensidad máxima y que pueden ser definidos según otra red, recíproca de la red directa anterior, produciéndose unas pautas o diseños típicos, como el que se produce cuando se observa un farol a través de un paraguas o una rendija.
 es el fenómeno físico a través del cual se manifiesta la interacción fundamental de los rayos X con los cristales (materia ordenada). Dispersión por un átomo. Llamamos factor atómico de dispersión (factor de scattering ) a la razón entre la amplitud dispersada por un átomo y la de un electrón aislado. Como la velocidad de los electrones en el átomo es mucho mayor que la variación del vector eléctrico de la onda, la radiación sólo ve una nube electrónica media, que viene caracterizada por la densidad electrónica de carga ρ(r). Si esta distribución se considera esféricamente simétrica, sólo depende de la distancia al núcleo. Dispersión por una red monoatómica: difracción. Cuando el agregado de átomos está estructurado según una red periódica tridimensional, de forma que los átomos constituyen nudos de esta red, las relaciones geométricas precisas entre los átomos del agregado dan lugar a diferencias de fase muy particulares. Se producen composiciones cooperativas entre las ondas dispersadas y la muestra actúa como una red de difracción de tres dimensiones. En estas condiciones, los efectos de interferencia externa producen efectos de dispersión estructurados en picos de intensidad máxima y que pueden ser definidos según otra red, recíproca de la red directa anterior, produciéndose unas pautas o diseños típicos, como el que se produce cuando se observa un farol a través de un paraguas o una rendija.")
13
Difracción por un cristal
Entonces, en las condiciones planteadas inicialmente, es decir, con un haz de rayos X monocromático y despolarizado, de ondas planas, formadas por rayos paralelos de un frente de ondas común, perpendicular al vector unitario de propagación s0 y que baña completamente a la muestra, el modelo cinemático de interacción indica que en la muestra se producen haces difractados en la dirección unitaria s con una intensidad: I(H) = Ie(H) IF(H) IL(H) en donde H es el vector de dispersión, Ie es la intensidad dispersada por un electrón, IL es el efecto de interferencia externa debido a la estructura tridimensional en red, e IF es el cuadrado del llamado factor de estructura, que viene a dar cuenta del efecto de interferencia interna debido a las relaciones de fase geométrica entre todos los átomos incluidos en la celdilla unidad. Concretamente, se llama factor de estructura F(H) a la onda resultante de la dispersión provocada por todos los átomos en una dirección.
 = Ie(H) IF(H) IL(H) en donde H es el vector de dispersión, Ie es la intensidad dispersada por un electrón, IL es el efecto de interferencia externa debido a la estructura tridimensional en red, e IF es el cuadrado del llamado factor de estructura, que viene a dar cuenta del efecto de interferencia interna debido a las relaciones de fase geométrica entre todos los átomos incluidos en la celdilla unidad. Concretamente, se llama factor de estructura F(H) a la onda resultante de la dispersión provocada por todos los átomos en una dirección.")
14
En la expresión de la intensidad total, I(H), las condiciones de máximo debido a la repetición en red, traen como consecuencia que: -El fenómeno de difracción en muestras cristalinas tiene carácter discreto, espectral. -Las direcciones y la repetición de la red que forma el espectro no dependen del factor de estructura, es decir, sólo dependen de la red directa. El conocimiento de estas direcciones permite conocer la forma y el tamaño de la celdilla unidad directa, que así controla la posición de los máximos de difracción. -La intensidad de los máximos depende del valor del factor de estructura en esa dirección, es decir, en ese punto recíproco, y estos máximos sólo dependen del contenido de la distribución atómica dentro de la celda unidad, que es lo que controla las intensidades de los máximos. Es decir, dan información sobre la estructura atómica dentro de la celda unidad. -El espectro total de difracción es el efecto de difracción del agregado atómico que forma la celda unidad muestreado en los puntos del espectro de difracción de la red cristalina que lo estructura. -En esencia, pues, la Cristalografía estructural por difracción de rayos X, consiste en medir las intensidades de la mayor cantidad posible de haces difractados del espectro tridimensional de difracción, obtener de ellas los módulos de los factores de estructura, y de sus valores, mediante algún procedimiento de asignación de fases a cada uno de estos factores, reconstruir la distribución electrónica en la celdilla elemental, cuyos máximos corresponderán a las posiciones atómicas.

15
Diagramas de difracción de (a) una molécula, (b) dos moléculas, (c) cuatro moléculas, (d) una línea de moléculas repetidas periódicamente, (e) dos líneas de moléculas y (f) una red bidimensional periódica de moléculas.
 una molécula, (b) dos moléculas, (c) cuatro moléculas, (d) una línea de moléculas repetidas periódicamente, (e) dos líneas de moléculas y (f) una red bidimensional periódica de moléculas.")
16
LEY DE BRAGG n λ = 2 dhkl sen θnh,nk,nl siendo n un número entero
Premio Nobel de Física 1915 n λ = 2 dhkl sen θnh,nk,nl siendo n un número entero La ecuación de Bragg tiene una interpretación sencilla, y es que cuando en la interacción cristal-radiación se produce una situación de máximo de difracción, el fenómeno es como si la radiación incidente se estuviera reflejando en la secuencia de planos cristalinos de índices hkl y espaciado dhkl. Es por eso por lo que, hablando de máximos de difracción, se use a veces la palabra reflexión de Bragg. Por otro lado, esta ecuación encierra las típicas relaciones de reciprocidad de la difracción, entre espaciado / dirección ó entre posición / momento: a menor espaciado, mayor ángulo y viceversa; redes directas con celdillas elementales grandes producen haces muy próximos, y al revés. La figura representa la descripción geométrica de la dirección del máximo de difracción debido a la interferencia constructiva entre los átomos de los planos de espaciado d(hkl). En la figura se da una descripción del modelo de Bragg cuando se trata de secuencias de planos del mismo espaciado, pero formados a su vez por átomos de distinto tipo, separados por Δd. Esta separación geométrica origina diferencias de fase dentro de un mismo haz difractado que provocan interferencias y que dan lugar a variaciones de intensidad (según la dirección), lo que permite obtener información de la estructura de los átomos que forman el cristal.
. En la figura se da una descripción del modelo de Bragg cuando se trata de secuencias de planos del mismo espaciado, pero formados a su vez por átomos de distinto tipo, separados por Δd. Esta separación geométrica origina diferencias de fase dentro de un mismo haz difractado que provocan interferencias y que dan lugar a variaciones de intensidad (según la dirección), lo que permite obtener información de la estructura de los átomos que forman el cristal.")
17
Difracción experimental
Las técnicas usadas para medir los ángulos y la intensidad de los haces de difracción han evolucionado a lo largo del tiempo. En el primer experimento de difracción, Friedrich y Knipping (1912) usaron una película sensible a los rayos X, pero incluso en el mismo año, Bragg usó una cámara de ionización montada sobre un brazo rotatorio que, en general, determinaba con más precisión los ángulos y las intensidades de difracción. Sin embargo, la película representó la ventaja de poder impresionar muchos haces difractados a la vez, y así durante los primeros años de la Cristalografía estructural (desde 1920 hasta 1970) se hizo uso extensivo de los métodos fotográficos, y entre ellos se deben destacar los métodos de Laue, Weissenberg, precesión y oscilación. A partir de mediados de la década de 1970, los métodos fotográficos fueron paulatinamente reemplazados por métodos goniométricos acoplados a detectores puntuales y posteriormente éstos últimos han sido reemplazados por detectores de área. Goniometría de cuatro círculos La introducción de los computadores digitales a finales de la década de 1970, permitió el diseño de los llamados difractómetros automáticos de cuatro círculos. Estos equipos, disponen de un sistema goniométrico, con una mecánica muy precisa, que mediante tres giros permite colocar el cristal en cualquier orientación del espacio, provocando así que se cumplan los requerimientos de la construcción de Ewald para que se produzca la difracción. En estas condiciones, un cuarto eje de giro, que sustenta el detector electrónico se coloca en condiciones de recoger el haz difractado. Todos estos movimientos se pueden programar para que se realicen de un modo automático, con una mínima intervención del operador. Esquema y aspecto de un goniómetro de cuatro círculos con geometría Kappa.
 usaron una película sensible a los rayos X, pero incluso en el mismo año, Bragg usó una cámara de ionización montada sobre un brazo rotatorio que, en general, determinaba con más precisión los ángulos y las intensidades de difracción. Sin embargo, la película representó la ventaja de poder impresionar muchos haces difractados a la vez, y así durante los primeros años de la Cristalografía estructural (desde 1920 hasta 1970) se hizo uso extensivo de los métodos fotográficos, y entre ellos se deben destacar los métodos de Laue, Weissenberg, precesión y oscilación. A partir de mediados de la década de 1970, los métodos fotográficos fueron paulatinamente reemplazados por métodos goniométricos acoplados a detectores puntuales y posteriormente éstos últimos han sido reemplazados por detectores de área. Goniometría de cuatro círculos La introducción de los computadores digitales a finales de la década de 1970, permitió el diseño de los llamados difractómetros automáticos de cuatro círculos. Estos equipos, disponen de un sistema goniométrico, con una mecánica muy precisa, que mediante tres giros permite colocar el cristal en cualquier orientación del espacio, provocando así que se cumplan los requerimientos de la construcción de Ewald para que se produzca la difracción. En estas condiciones, un cuarto eje de giro, que sustenta el detector electrónico se coloca en condiciones de recoger el haz difractado. Todos estos movimientos se pueden programar para que se realicen de un modo automático, con una mínima intervención del operador. Esquema y aspecto de un goniómetro de cuatro círculos con geometría Kappa.")
18
Detectores de área Como alternativa a los detectores "puntuales", el desarrollo de la tecnología electrónica ha dado lugar a la aparición de los denominados detectores de área, que permiten la detección de muchos haces de difracción simultáneamente, ahorrando con ello tiempo en el experimento. Esta tecnología es de especial utilidad para el caso de las proteínas y en general de cualquier material que pueda deteriorarse durante su exposición a los rayos X, ya que la detección de cada una de las imágenes que se recogen (con varios cientos o miles de reflexiones) se hace en un tiempo mínimo, del orden de los segundos. Uno de los detectores de área mas comunmente usado se basa en los denominados CCD's (del inglés Charge Coupled Device) cuyo esquema se muestra a continuación:
 se hace en un tiempo mínimo, del orden de los segundos. Uno de los detectores de área mas comunmente usado se basa en los denominados CCD s (del inglés Charge Coupled Device) cuyo esquema se muestra a continuación:")
19
Resolución estructural
Los electrones en los átomos (y por lo tanto las moléculas) que están en los cristales (ordenados) dispersan los rayos X de modo peculiar, cooperativo, que denominamos difracción Las ondas dispersadas de este modo cooperativo se componen a su vez, en cascada, unas con otras, dando lugar a ondas totales resultantes en cada dirección de difracción, de tal modo que, dependiendo del "defase" de unas con respecto a otras, éstas se suman o se restan, tal como se muestra en la figura de abajo: Composición de dos ondas. A = amplitud resultante; I = intensidad resultante (∼ A2) (a) totalmente en fase (el efecto total es la suma de ambas) (b) con un cierto defase (hay suma pero no total) (c) en oposición de fase (la resultante es nula)
 que están en los cristales (ordenados) dispersan los rayos X de modo peculiar, cooperativo, que denominamos difracción. Las ondas dispersadas de este modo cooperativo se componen a su vez, en cascada, unas con otras, dando lugar a ondas totales resultantes en cada dirección de difracción, de tal modo que, dependiendo del defase de unas con respecto a otras, éstas se suman o se restan, tal como se muestra en la figura de abajo: Composición de dos ondas. A = amplitud resultante; I = intensidad resultante (∼ A2) (a) totalmente en fase (el efecto total es la suma de ambas) (b) con un cierto defase (hay suma pero no total) (c) en oposición de fase (la resultante es nula)")
20
LA DENSIDAD ELECTRÓNICA
Conocer (ó ver) la estructura interna de un cristal supone poder resolver una función matemática que define la denominada "densidad electrónica", que es una función que está definida en cada punto de la celda unidad. Esta función de densidad electrónica, representada por la letra griega ρ tiene un valor determinado en cada punto (x, y, z) de la celda elemental y allí en donde toma valores máximos (estimados en términos de "electrones por Angstrom cúbico") es en donde estarán localizados los átomos que componen un cristal, dándonos por lo tanto la localización de los mismos. F(hkl) representa a las ondas resultantes de la dispersión de todos los átomos en cada una de las direcciones y se denominan factores de estructura. Sus módulos están directamente relacionados con las intensidades de las reflexiones del espectro) h, k, l son los índices de Miller de las reflexiones y Φ(hkl) representa las denominadas "fases" de las reflexiones (las fases de unas ondas respecto de otras) Aspecto de una zona del mapa de densidad electrónica de un cristal de proteína, antes de su interpretación El mismo mapa de densidad electrónica de la izquierda interpretado en términos de un fragmento peptídico
 la estructura interna de un cristal supone poder resolver una función matemática que define la denominada densidad electrónica , que es una función que está definida en cada punto de la celda unidad. Esta función de densidad electrónica, representada por la letra griega ρ tiene un valor determinado en cada punto (x, y, z) de la celda elemental y allí en donde toma valores máximos (estimados en términos de electrones por Angstrom cúbico ) es en donde estarán localizados los átomos que componen un cristal, dándonos por lo tanto la localización de los mismos. F(hkl) representa a las ondas resultantes de la dispersión de todos los átomos en cada una de las direcciones y se denominan factores de estructura. Sus módulos están directamente relacionados con las intensidades de las reflexiones del espectro) h, k, l son los índices de Miller de las reflexiones y Φ(hkl) representa las denominadas fases de las reflexiones (las fases de unas ondas respecto de otras) Aspecto de una zona del mapa de densidad electrónica de un cristal de proteína, antes de su interpretación. El mismo mapa de densidad electrónica de la izquierda interpretado en términos de un fragmento peptídico.")
21
EL PROBLEMA DE LAS FASES
Sin embargo, para poder calcular la función de densidad electrónica (ρ(xyz)), y por lo tanto poder saber la localización de los átomos en el interior de la celda, necesitamos conocer también el defase entre las ondas (variable Φ(hkl) en la ecuación de la densidad electrónica) y esta información "se nos escapa" durante el proceso de medida experimental, ya que no existen técnicas experimentales para medir esos defases! El problema está, pues, servido y esta dificultad es la que ha dado nombre al concepto del problema de las fases. Hay distintos métodos para resolver esto: Patterson, directos, MIR, etc). LA FUNCIÓN DE PATTERSON Basándose en la imposibilidad de resolver de un modo directo la función de la densidad electrónica Patterson introdujo una nueva función P(uvw) que supone una simplificación de la información contenida en la función de densidad electrónica, ya que suprime la información de las fases, y los módulos de los factores de estructura se sustituyen por sus cuadrados. Es, pues, una función que puede calcularse de inmediato a partir de la información experimental de que se dispone (las intensidades, que a su vez se derivan de los módulos de los factores de estructura). P(uvw) = (1/V) ΣΣΣ [F(hkl)]2 cos 2π (hu + kv + lw)) La información que proporciona la función de Patterson es un mapa de vectores de posición entre átomos (posiciones relativas). Los máximos de la función de Patterson son tanto más altos cuanto mayores sean los números de electrones de los átomos implicados, lo cual supone una gran ventaja en el caso de moléculas que contengan especies de número atómico alto. Calculada dicha la función, P(uvw), se trata pues de interpretarla correctamente (obtener la posición absoluta de algunos átomos) para, indirectamente, alcanzar el conocimiento necesario, fases Φ(hkl), que nos permitan visualizar la función de la densidad electrónica ρ(xyz).
), y por lo tanto poder saber la localización de los átomos en el interior de la celda, necesitamos conocer también el defase entre las ondas (variable Φ(hkl) en la ecuación de la densidad electrónica) y esta información se nos escapa durante el proceso de medida experimental, ya que no existen técnicas experimentales para medir esos defases! El problema está, pues, servido y esta dificultad es la que ha dado nombre al concepto del problema de las fases. Hay distintos métodos para resolver esto: Patterson, directos, MIR, etc). LA FUNCIÓN DE PATTERSON. Basándose en la imposibilidad de resolver de un modo directo la función de la densidad electrónica Patterson introdujo una nueva función P(uvw) que supone una simplificación de la información contenida en la función de densidad electrónica, ya que suprime la información de las fases, y los módulos de los factores de estructura se sustituyen por sus cuadrados. Es, pues, una función que puede calcularse de inmediato a partir de la información experimental de que se dispone (las intensidades, que a su vez se derivan de los módulos de los factores de estructura). P(uvw) = (1/V) ΣΣΣ [F(hkl)]2 cos 2π (hu + kv + lw)) La información que proporciona la función de Patterson es un mapa de vectores de posición entre átomos (posiciones relativas). Los máximos de la función de Patterson son tanto más altos cuanto mayores sean los números de electrones de los átomos implicados, lo cual supone una gran ventaja en el caso de moléculas que contengan especies de número atómico alto. Calculada dicha la función, P(uvw), se trata pues de interpretarla correctamente (obtener la posición absoluta de algunos átomos) para, indirectamente, alcanzar el conocimiento necesario, fases Φ(hkl), que nos permitan visualizar la función de la densidad electrónica ρ(xyz).")
22
COMPLETANDO LA ESTRUCTURA
Todos estos métodos (Patterson, métodos directos, MIR, MAD, MR) proporcionan (directa o indirectamente) un conocimiento aproximado de las fases, lo que denominamos fases iniciales, que hay que mejorar. Tal como se indica en el párrafo anterior, estas fases iniciales calculadas, Φc(hkl), junto con las amplitudes experimentales observadas, |Fo(hkl)|, nos permitirán calcular una función de densidad electrónica, también aproximada, y sobre la cual podremos construir nuestro modelo estructural. El proceso general a seguir se resume, cíclicamente, en el esquema siguiente. Las fases iniciales, Φc(hkl), se pueden combinar con los módulos de los factores de estructura experimentales (observados) |Fo(hkl)| para calcular la función de densidad electrónica (parte inferior del esquema). Alternativamente, si el conocimiento inicial han sido las posiciones atómicas (xyz) de algunos átomos, éstos proporcionarán las fases iniciales (parte superior del esquema). A partir de las posiciones atómicas conocidas (xyz) se pueden obtener los módulos de factores de estructura y fases calculados, |Fc(hkl)| y Φc(hkl) (parte superior), pero es obvio que los factores de estructura calculados sean rechazados por estar evaluados con una estructura parcial y los experimentales son consecuencia de la estructura completa. Así pues, la función de densidad electrónica se calcula con los módulos experimentales de los factores de estructura (los observados, |Fo(hkl)|) y las fases calculadas Φc(hkl). Dicha función se evalua en términos de la posible información añadida que proporcione y el ciclo se repite hasta que no se obtenga información adicional. Históricamente este proceso se conoció como "síntesis sucesivas de Fourier" ya que la función de densidad electrónica, que en realidad es una integral, se calcula como una suma de Fourier. Esquema de los cálculos sucesivos de un mapa de densidad electrónica, ρ(xyz).
 proporcionan (directa o indirectamente) un conocimiento aproximado de las fases, lo que denominamos fases iniciales, que hay que mejorar. Tal como se indica en el párrafo anterior, estas fases iniciales calculadas, Φc(hkl), junto con las amplitudes experimentales observadas, |Fo(hkl)|, nos permitirán calcular una función de densidad electrónica, también aproximada, y sobre la cual podremos construir nuestro modelo estructural. El proceso general a seguir se resume, cíclicamente, en el esquema siguiente. Las fases iniciales, Φc(hkl), se pueden combinar con los módulos de los factores de estructura experimentales (observados) |Fo(hkl)| para calcular la función de densidad electrónica (parte inferior del esquema). Alternativamente, si el conocimiento inicial han sido las posiciones atómicas (xyz) de algunos átomos, éstos proporcionarán las fases iniciales (parte superior del esquema). A partir de las posiciones atómicas conocidas (xyz) se pueden obtener los módulos de factores de estructura y fases calculados, |Fc(hkl)| y Φc(hkl) (parte superior), pero es obvio que los factores de estructura calculados sean rechazados por estar evaluados con una estructura parcial y los experimentales son consecuencia de la estructura completa. Así pues, la función de densidad electrónica se calcula con los módulos experimentales de los factores de estructura (los observados, |Fo(hkl)|) y las fases calculadas Φc(hkl). Dicha función se evalua en términos de la posible información añadida que proporcione y el ciclo se repite hasta que no se obtenga información adicional. Históricamente este proceso se conoció como síntesis sucesivas de Fourier ya que la función de densidad electrónica, que en realidad es una integral, se calcula como una suma de Fourier. Esquema de los cálculos sucesivos de un mapa de densidad electrónica, ρ(xyz).")
23
AJUSTE FINAL DEL MODELO
El modelo estructural El análisis de la función de densidad electrónica, es decir, la resolución de una estructura cristalina (molecular, o no molecular) nos conduce a un modelo inicial, no definitivo, que se describe por las posiciones relativas de los átomos, los cuales pueden representarse mediante puntos o pequeñas esferas: AJUSTE FINAL DEL MODELO La obtención de este modelo "ajustado" (refinado) es consecuencia de metodología matemática analítica como la de los mínimos cuadrados. Mediante esta técnica se "mueven" ligeramente las posiciones atómicas (las coordenadas) y se aplican factores térmicos a cada átomo de tal modo que el espectro de difracción calculado con dicho modelo sea prácticamente igual que el experimental (observado), es decir que se minimizan las diferencias entre los factores de estructura observados y calculados. Este proceso se lleva a cabo minimizando (haciendo tender a cero) la función: Σ w | |Fo| - |Fc| |2 → 0 en donde w representa un factor de "peso" asignado a cada observación, separando así los efectos de aquellas observaciones más precisas de las menos precisas y evitando así errores sistemáticos en las observaciones experimentales que pudieran sesgar el modelo.
 nos conduce a un modelo inicial, no definitivo, que se describe por las posiciones relativas de los átomos, los cuales pueden representarse mediante puntos o pequeñas esferas: AJUSTE FINAL DEL MODELO. La obtención de este modelo ajustado (refinado) es consecuencia de metodología matemática analítica como la de los mínimos cuadrados. Mediante esta técnica se mueven ligeramente las posiciones atómicas (las coordenadas) y se aplican factores térmicos a cada átomo de tal modo que el espectro de difracción calculado con dicho modelo sea prácticamente igual que el experimental (observado), es decir que se minimizan las diferencias entre los factores de estructura observados y calculados. Este proceso se lleva a cabo minimizando (haciendo tender a cero) la función: Σ w | |Fo| - |Fc| |2 → 0. en donde w representa un factor de peso asignado a cada observación, separando así los efectos de aquellas observaciones más precisas de las menos precisas y evitando así errores sistemáticos en las observaciones experimentales que pudieran sesgar el modelo.")
24
VALIDACIÓN DEL MODELO Existen una serie de herramientas que ayudan a evaluar la fiabilidad de un modelo estructural, y que en términos cristalográficos se conocen con el nombre de validación, de tal modo que el modelo estructural obtenido debe ser contínuamente contrastado y validado mediante criterios estereoquímicos consistentes. Es decir, las distancias interatómicas y ángulos de enlace deben ser aceptables. No lo sería, por ejemplo, una distancia C---O de 0.8 Angström para un supuesto grupo carbonilo (C=O). Y del mismo modo, los ángulos de enlace, deben de ser consistentes con una geometría aceptable. Estos criterios son muy restrictivos para los modelos estructurales de compuestos de hasta mediana complejidad, pero incluso en las estructuras de las macromoléculas deben de cumplirse unos mínimos
. Y del mismo modo, los ángulos de enlace, deben de ser consistentes con una geometría aceptable. Estos criterios son muy restrictivos para los modelos estructurales de compuestos de hasta mediana complejidad, pero incluso en las estructuras de las macromoléculas deben de cumplirse unos mínimos.")
25
Esquema general que ilustra el proceso de resolución de estructuras moleculares y cristalinas mediante la difracción de rayos X

Presentaciones similares





>")














