Descargar la presentación
La descarga está en progreso. Por favor, espere

1
INTRODUCCIÓN A LA TERMODINÁMICA y a la TERMOQUÍMICA
Energía interna, calor y trabajo

2
Termodinámica Energía es la capacidad de producir trabajo o más ajustadamente LA ENERGÍA SE RELACIONA CON LA CAPACIDAD DE PRODUCIR TRABAJO, lo cual prefigura un concepto más amplio y adecuado Termodinámica es el estudio de las transformaciones e intercambios de la energía, entendida esta como la capacidad de un sistema para realizar un trabajo o para suministrar calor.

3
. Es imposible realizar un trabajo sin consumir una energía W=F x
LA ENERGÍA DEL UNIVERSO SE CONSERVA PRIMER PRINCIPIO Es imposible realizar un trabajo sin consumir una energía . uff, uff Fuerza distancia X1 X2 Trabajo=área [N.m=J] W=F x Trabajo realizado por el hombre Fuerza aplicada Distancia que se desplaza el objeto Energía = Capacidad para realizar un trabajo

4
LA ENERGÍA DEL UNIVERSO SE CONSERVA PRIMER PRINCIPIO
La pérdida de energía potencial acelera el deslizamiento del objeto cae se acelera La energía potencial se transforma en energía cinética Reacción Química Cambio de Fase energía química (carbón) energía interna (agua líquida vapor de agua) el vapor se expande Trabajo energía cinética
 energía interna (agua líquida vapor de agua) el vapor se expande Trabajo. energía cinética.")
5
Principio de Conservación de la energía
Todas las formas de energía tienden en última instancia a pasar a calor. Ejs. Energía química a calor, en una reacción química (más adelante veremos que esto no es tan general). Energía eléctrica Calor, cuando actúa a través de una resistencia, etc. LA ENERGÍA SE CONSERVA. Esto es más conocido como Principio de Conservación de la energía
. Energía eléctrica Calor, cuando actúa a través de una resistencia, etc. LA ENERGÍA SE CONSERVA. Esto es más conocido como Principio de Conservación de la energía.")
6
Sistemas termodinámicos
Definimos sistema como la "porción delimitada del mundo físico (y especificada) que contiene cantidades definidas de sustancia que se consideran bajo estudio"
 que contiene cantidades definidas de sustancia que se consideran bajo estudio")
7
Sistema y entorno ENTORNO es la zona del universo que interactúa con el sistema. SISTEMA ENERGÍA Sistema + Entorno (O Medio Ambiente) = UNIVERSO
 = UNIVERSO.")
8
Tipos de sistema Aislado: no hay transferencia de masa o energía con el entorno Ej. : un termo ideal (aislado y de paredes rígidas). Termo de café, calorímetro Cerrado: no transfiere masa pero sí energía en forma de calor, trabajo o radiación. Ej. : cualquier recipiente cerrado no ideal. Fiambrera cerrada Abierto: transfiere masa y energía con su entorno. Ej. : el cuerpo humano, Cilindro de un automóvil La mayoría de los sistemas en la vida real son abiertos, mientras que en el laboratorio la mayoría de los sistemas químicos son cerrados
. Termo de café, calorímetro. Cerrado: no transfiere masa pero sí energía en forma de calor, trabajo o radiación. Ej. : cualquier recipiente cerrado no ideal. Fiambrera cerrada. Abierto: transfiere masa y energía con su entorno. Ej. : el cuerpo humano, Cilindro de un automóvil. La mayoría de los sistemas en la vida real son abiertos, mientras que en el laboratorio la mayoría de los sistemas químicos son cerrados.")
9
Sistemas químicos

10
PRIMERA LEY DE LA TERMODINÁMICA
La Ley de Conservación de la Energía es una de las formas de expresión de la Primera Ley de la Termodinámica. Se puede visualizar en una forma muy sencilla con los conceptos de ENERGÍA ACUMULADA y ENERGÍA INTERCAMBIADA O EN PROCESO DE INTERCAMBIO o también ENERGÍA EN TRÁNSITO

11
Es final ‑ Es inicial = SEi o lo que es igual
SEs = SEI

12
Por otra parte si bien hay un INTERCAMBIO de Energía entre Sistema‑Medio Ambiente siempre se cumple la LEY DE CONSERVACIÓN DE LA ENERGÍA ya que: 1. ‑ ANTES DEL INTERCAMBIO: Esi + Emai = Euniverso siendo i= inicial 2. ‑ LUEGO DEL INTERCAMBIO: Esf + Emaf = Euniverso siendo f= final DONDE: DEs = Esf ‑ Esi = SEs = SEintercambiadas En ambos casos la ENERGÍA DEL UNIVERSO se mantiene constante

13
ENERGÍA INTERNA La Energía Interna (E) se define en la bibliografía como TODAS LAS FORMAS DE ENERGÍA DE UN SISTEMA, DISTINTAS DE LAS QUE RESULTEN DE SU POSICIÓN EN EL ESPACIO (Energía potencial, que se supone constante). Nosotros además agregamos que también SE DEBE DESCARTAR LA ENERGÍA CINÉTICA GLOBAL DEL SISTEMA
 se define en la bibliografía como TODAS LAS FORMAS DE ENERGÍA DE UN SISTEMA, DISTINTAS DE LAS QUE RESULTEN DE SU POSICIÓN EN EL ESPACIO (Energía potencial, que se supone constante). Nosotros además agregamos que también SE DEBE DESCARTAR LA ENERGÍA CINÉTICA GLOBAL DEL SISTEMA.")
14
Energías que componen la energía interna
ENERGÍAS INTRAMOLECULARES 1. Nuclear 2. Electrónica 3. Traslacional 4. Vibracional 5. Rotacional (3, 4, y 5 son las Energías Térmicas)
")
15
Energías que componen la energía interna
ENERGÍAS INTERMOLECULARES Fuerzas de atracción interiónicas (sales) Fuerzas de atracción ion‑dipolo Unión por puente de hidrógeno Fuerzas de atracción dipolo‑dipolo Fuerzas de atracción entre iones o dipolos‑moléc.polarizables Fuerzas entre dipolos instantáneos‑dip. inducidos (London) Fuerzas hidrofóbicas Fuerzas de repulsión
 Fuerzas de atracción ion‑dipolo. Unión por puente de hidrógeno. Fuerzas de atracción dipolo‑dipolo. Fuerzas de atracción entre iones o dipolos‑moléc.polarizables. Fuerzas entre dipolos instantáneos‑dip. inducidos (London) Fuerzas hidrofóbicas. Fuerzas de repulsión.")
16
El valor de la energía interna
NO ES POSIBLE CALCULAR EL VALOR ABSOLUTO DE LA ENERGÍA INTERNA ¿QUÉ PODEMOS CALCULAR? LOS CAMBIOS DE ENERGÍA INTERNA

17
Calor Energía que se transfiere de un objeto a otro debido a una diferencia de temperatura C = [J/ºK] cal=4.184 J Una caloría es el calor necesario para elevar la temperatura de 1g de agua 1ºC Calor específico molar Calor específico Capacidad Calorífica
![Calor Energía que se transfiere de un objeto a otro debido a una diferencia de temperatura. C = [J/ºK] 1cal=4.184 J.](http://slideplayer.es/slide/5433484/17/images/17/Calor+Energ%C3%ADa+que+se+transfiere+de+un+objeto+a+otro+debido+a+una+diferencia+de+temperatura.+C+%3D+%5BJ%2F%C2%BAK%5D+1cal%3D4.184+J..jpg "Una caloría es el calor necesario para elevar la temperatura de 1g de agua 1ºC. Calor específico. molar. Calor. específico. Capacidad. Calorífica.")
18
Cambios de fase Cambio de fase y calor latente
Calor de fusión = Calor necesario para fundir una sustancia sin modificar su temperatura. Calor de evaporación = Calor necesario para vaporizar una sustancia sin modificar su temperatura.

19
Calor. Convenio de signos
Sistema Q<0 Q>0 Calor cedido por el sistema Calor absorbido por el sistema ENDOTÉRMICO POSITIVO (+) EXOTÉRMICO NEGATIVO (-)
 EXOTÉRMICO NEGATIVO (-)")
20
Trabajo como forma de intercambio de energía.
Unidades TRABAJO (PV) Pext > Pint Pext Pint dx Pext Pint Pext = Pint Estado inicial Equilibrio mecánico Estado final Pext = Pint
 Pext > Pint. Pext. Pint. dx. Pext. Pint. Pext = Pint. Estado. inicial. Equilibrio mecánico. Estado. final. Pext = Pint.")
21
Trabajo Ejemplo: gas expansionado contra un pistón móvil w = F x d = (P x A) x h = PV w = -PextV w = - Pext . DV

22
Trabajo como forma de intercambio de energía
Convención de signos para TRABAJO PROCESO SIGNO SOBRE EL SISTEMA POSITIVO (+) POR EL SISTEMA NEGATIVO (-)
 POR EL SISTEMA NEGATIVO (-)")
23
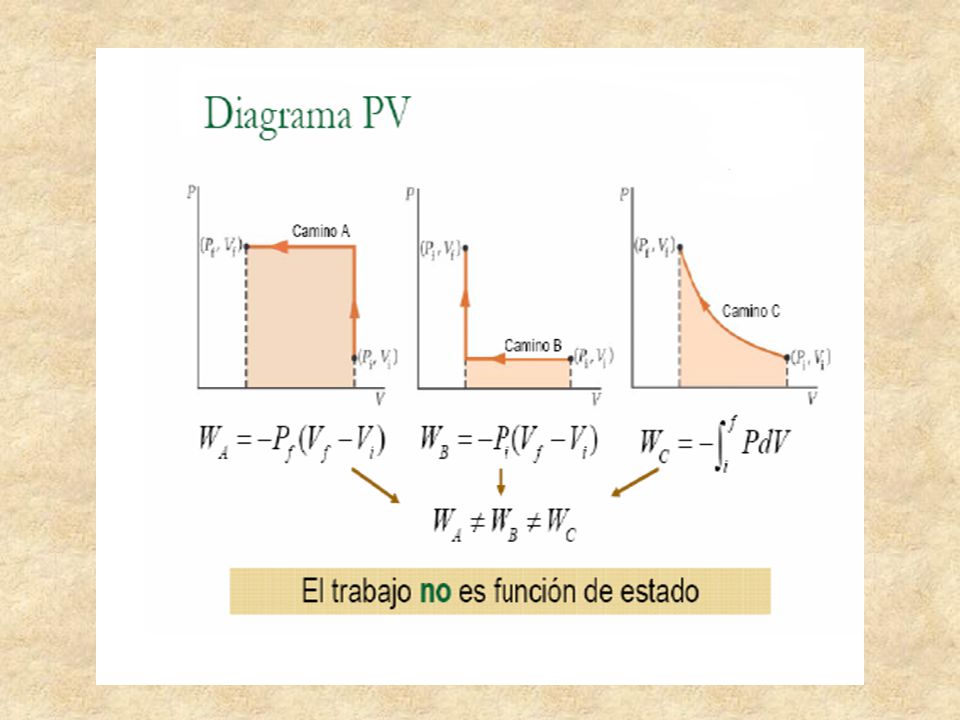
Trabajo como forma de intercambio de energía
El trabajo efectuado en la expansión desde el estado inicial hasta el estado final es el área bajo la curva en un diagrama PV. Proceso casi estático significa que el proceso ocurre lentamente y en todo momento el sistema esta en equilibrio.

24
¿El proceso puede ser reversible?
Sí, siempre qué: Pext = Pinterna o mejor: Pext = Pinterna + dP dP = diferencial de presión o sea un incremento de presión infinitesimal Sólo entonces cuando ello ocurra estaremos en presencia de un PROCESO REVERSIBLE TERMODINAMICAMENTE el cual en todo momento debe estar en EQUILIBRIO.

25
PARA UN PROCESO ISÓBARO
= - P (Vf-Vi) W Pext < Pint Pext Pint Pext Pint Pext = Pint V V i Vf
 W. Pext < Pint. Pext. Pint. Pext. Pint. Pext = Pint. V. V i Vf.")
26
PROCESOS ISOTERMOS Supongamos un gas ideal P0V0 = nRT P 1 V 1 = nR T
Son aquellos que se realizan a T=cte Supongamos un gas ideal P0V0 = nRT P 1 V 1 = nR T Entonces:

27
Expresión general del trabajo
Esta expresión es válida para un camino ISOTÉRMICO y REVERSIBLE EN ESAS CONDICIONES EL TRABAJO ES EL MÁXIMO POSIBLE o TRABAJO MÁXIMO Cualquier otro proceso no logrará una cantidad de trabajo similar

28
FUNCIÓN DE ESTADO En termodinámica, la descripción del estado de un sistema se realiza mediante los valores de determinadas propiedades macroscópicas denominadas variables termodinámicas, tales como p, V, T, m, ... No todas estas variables son independientes, basta conocer los valores de un pequeño número de ellas para caracterizar el sistema. Estas variables independientes se denominan variables de estado. Toda función que pueda expresarse con ayuda de las variables de estado se denomina función de estado del sistema.

29
FUNCIÓN DE ESTADO Una función de estado es cualquier propiedad que tiene un único valor cuando el estado del sistema está definido Es aquella cuyo resultado no dependa del camino seguido sino del estado final y del inicial del sistema en estudio Una muestra de agua a 293,15 K y a la presión de una atmósfera está en un estado especificado. d = 0,99820 g/mL. Esta densidad es una función única del estado. No importa cómo se haya establecido el sistema.

30
Extensivas Intensivas Tipos de variables
Los sistemas se presentan de diferentes formas Þ ESTADOS caracterizados por VARIABLES termodinámicas Variable = Propiedad Termodinámica = Función de Estado No dependen de la historia Extensivas Intensivas Tipos de variables No dependen de la cantidad de materia del sistema Ej: T, P, r No son aditivas Dependen de la cantidad Ej: m, V Son aditivas

32
Trabajo “no-útil” y “útil”

33
Q > 0 W > 0 W < 0 Q < 0 TRABAJO. CALOR, ENERGÍA.
Criterio de signos SISTEMA Q > 0 W > 0 W < 0 Q < 0

34
Expresión matemática de la Primera Ley (Cálculo de la variación de Energía interna (E) de un sistema) Al ocurrir un cambio de energías (calor y trabajo, o una sola de ellas) la variación de energía del sistema (que es el que interesa), en definitiva es la variación de DE que se busca conocer: DEs = DEI
 la variación de energía del sistema (que es el que interesa), en definitiva es la variación de DE que se busca conocer: DEs = DEI.")
35
T(i) = 20°C T(f) = 21°C T(i) = 20°C T(f) = 21°C
q = 999,43cal w= q=0 w = +4181,6 J = +999,43 cal DE = q + (-)w = 999,43 cal DE = ,43 cal = 999,43 cal De aquí surgen dos hechos inmediatos: a.‑ E (energía interna) es una FUNCIÓN DE ESTADO. b.- q (calor) en un caso tiene un valor finito y en el otro vale cero por que los caminos son distintos, y lo mismo ocurre con w (trabajo). El calor y el trabajo dependen necesariamente del CAMINO que se sigue y por lo tanto NO SON FUNCIONES DE ESTADO.
w = 999,43 cal DE = ,43 cal = 999,43 cal. De aquí surgen dos hechos inmediatos: a.‑ E (energía interna) es una FUNCIÓN DE ESTADO. b.- q (calor) en un caso tiene un valor finito y en el otro vale cero por que los caminos son distintos, y lo mismo ocurre con w (trabajo). El calor y el trabajo dependen necesariamente del CAMINO que se sigue y por lo tanto NO SON FUNCIONES DE ESTADO.")
36
Primer principio de la Termodinámica
El calor añadido a un sistema es igual a la variación de energía interna del mismo más el trabajo realizado por el sistema Variaciones infinitesimales

37
1er Principio de la Termodinámica
2.- PRIMER PRINCIPIO. ENERGÍA INTERNA. ENERGÍA No es posible conocer la energía de un sistema, sólo conocemos su cambio en un proceso U=U2-U1 Energía interna (U) (Suma de energías a nivel molecular) Función de estado Magnitud extensiva ¿Cómo podemos aumentar U de un sistema cerrado? Realizando un trabajo Calentándolo Þ calor DU = Q + W 1er Principio de la Termodinámica
 (Suma de energías a nivel molecular) Función de estado. Magnitud extensiva. ¿Cómo podemos aumentar U. de un sistema cerrado Realizando un trabajo. Calentándolo Þ calor. DU = Q + W. 1er Principio de la Termodinámica.")
38
Aplicaciones del primer Principio. Proceso isócoro
V= cte p P2 P1 V

39
Aplicaciones del primer Principio. Proceso isóbaro
Isóbara P=cte p V V1 V2 n = 1

40
PROCESOS ISOTERMOS Como no hay variación de temperatura
NO habrá variación de ENERGÍA INTERNA El proceso isotermo es un proceso claramente REVERSIBLE

41
Proceso isotermo (Gas ideal)
T =cte p V2 V V1

42
PROCESO termodinámico
Trayectoria = Camino que sigue el sistema cuando su estado , las funciones de estado, cambia con el tiempo ß PROCESO termodinámico Tipos de procesos Isotermo (T = cte) Isobaro (P = cte) Isocoro (V = cte) Adiabático (Q = 0) Cíclico (estado final = estado inicial) Irreversible Reversible (sistema siempre infinitesimalmente próximo al equilibrio; un cambio infinitesimal en las condiciones puede invertir el proceso) (un cambio infinitesimal en las condiciones no produce un cambio de sentido en la transformación).
 Isobaro (P = cte) Isocoro (V = cte) Adiabático (Q = 0) Cíclico (estado final = estado inicial) Irreversible. Reversible. (sistema siempre infinitesimalmente próximo al equilibrio; un cambio infinitesimal en las condiciones puede invertir el proceso) (un cambio infinitesimal en las condiciones no produce un cambio de sentido en la transformación).")
43
Proceso Cíclico Es un proceso que empieza y termina en el mismo estado. En este caso el cambio de energía interna es cero y el calor agregado al sistema debe ser igual al trabajo realizado durante el ciclo, entonces: U = y Q = W Expansión contra el vació. Si la presión externa es cero, de la expresión se deduce que W = 0. Durante la expansión libre no se realiza trabajo. Si el proceso es isotérmico, U = 0 por ende Q = W = 0 w = - Pext . DV

44
1er Principio de la Termodinámica
PRIMER PRINCIPIO. ENERGÍA INTERNA. DU = Q + W 1er Principio de la Termodinámica Proceso Cíclico A→A A P B Es imposible realizar un trabajo sin consumir una energía V

45
1er Principio de la Termodinámica
PRIMER PRINCIPIO. ENERGÍA INTERNA. DU = Q + W 1er Principio de la Termodinámica Proceso Cíclico A→A A P U función de estado B Es imposible realizar un trabajo sin consumir una energía V

46
Trabajo “no-útil” y “útil”
Patm . V = Trabajo de expansión contra la P atm. NO ÚTIL. p. V = Trabajo de expansión contra la Presión "extra" que arbitrariamente hemos colocado: este es un trabajo de EXPANSIÓN ÚTIL ya que la pesa (p) queda al final del proceso con una cierta energía potencial mayor al final que al comienzo del proceso. ¿Esto es común en nuestros cálculos? No lo es. Por ello, y en general el trabajo de expansión será no-útil.
 queda al final del proceso con una cierta energía potencial mayor al final que al comienzo del proceso. ¿Esto es común en nuestros cálculos No lo es. Por ello, y en general el trabajo de expansión será no-útil.")
47
Expresión general del trabajo:
w=(P‑dP) dV = P dV ‑ dp.dV donde dp.dV se desprecia por ser un diferencial de orden superior dw = P . dV y de acuerdo a la convención adoptada será dw = - P . dV
 dV = P dV ‑ dp.dV. donde dp.dV se desprecia por ser un diferencial de orden superior. dw = P . dV. y de acuerdo a la convención adoptada será dw = - P . dV.")
48
Expresión general del trabajo:
Se integra la ecuación y se llega a una expresión tal como

49
Diferentes formas de trabajo
a) Eléctrico b) Osmótico c) Luz d) Químico e) Muscular f) Gravitacional g) De superficie
 Eléctrico. b) Osmótico. c) Luz. d) Químico. e) Muscular. f) Gravitacional. g) De superficie.")
50
Primera ley o Primer Principio
Esta sumatoria de energías intercambiadas, es como su nombre lo indica DEI = calor + trabajo. Esto es, la energía en forma de calor que se ganó o perdió más la energía en forma de trabajo que se ganó o perdió

51
Primera ley o Primer Principio: caso 1
RECIBIÓ CALOR: AUMENTÓ SU ENERGÍA INTERNA REALIZÓ UN TRABAJO (por el sistema): PERDIÓ ENERGÍA INTERNA
: PERDIÓ ENERGÍA INTERNA.")
52
Primera ley o Primer Principio: caso 1
Luego para conocer en cuánto varió la ENERGÍA INTERNA (E) del sistema debemos calcular la sumatoria de ENERGÍAS INTERCAMBIADAS, pero para ello debe recordarse la convención de signos, donde el Calor es positivo y el Trabajo (de expansión) que según el cálculo es positivo, debe cambiarse su signo a negativo; luego la sumatoria será: SEI = SEs = E = q + w
 del sistema debemos calcular la sumatoria de ENERGÍAS INTERCAMBIADAS, pero para ello debe recordarse la convención de signos, donde el Calor es positivo y el Trabajo (de expansión) que según el cálculo es positivo, debe cambiarse su signo a negativo; luego la sumatoria será: SEI = SEs = E = q + w.")
53
Primera ley o Primer Principio: caso 2
CEDIÓ CALOR (EXOTÉRMICO), LUEGO DISMINUYÓ SU ENERGÍA INTERNA RECIBIÓ UN TRABAJO (sobre el sistema): AUMENTÓ SU ENERGÍA INTERNA
, LUEGO DISMINUYÓ SU ENERGÍA INTERNA. RECIBIÓ UN TRABAJO (sobre el sistema): AUMENTÓ SU ENERGÍA INTERNA.")
54
Primera ley o Primer Principio: caso 2
Por lo tanto: SEI = DE = q + w donde "q" debe mantenerse con el signo de acuerdo al proceso y "w" debe sumarse ya que aumenta la E

55
Primera ley o Primer Principio: caso 3
CEDIÓ CALOR (EXOTÉRMICO) LUEGO PERDIÓ ENERGÍA INTERNA REALIZÓ UN TRABAJO (POR EL SISTEMA) LUEGO PERDIÓ ENERGÍA INTERNA
 LUEGO PERDIÓ ENERGÍA INTERNA. REALIZÓ UN TRABAJO (POR EL SISTEMA) LUEGO PERDIÓ ENERGÍA INTERNA.")
56
Primera ley o Primer Principio: caso 3
Por lo tanto: SEI = DE = q + w

57
Primera ley o Primer Principio: caso 4
RECIBIÓ CALOR (proceso endotérmico) AUMENTANDO SU ENERGÍA RECIBIÓ UN TRABAJO (sobre el sistema) AUMENTANDO SU ENERGÍA
 AUMENTANDO SU ENERGÍA. RECIBIÓ UN TRABAJO (sobre el sistema) AUMENTANDO SU ENERGÍA.")
58
Primera ley o Primer Principio: caso 4
Por lo tanto: SEI = DE = q + w

59
EXPRESIÓN DE LA PRIMERA LEY CONSIDERANDO LOS TRABAJOS POSIBLES
DE = SEI = DEs = q + wexp + wútil Esta expresión no se usa, al menos en los cursos introductorios de Termodinámica SIEMPRES SE USA ESTA EXPRESIÓN DE = SEI = DEs= q + wexp

60
Energía interna de un gas ideal
Única energía existente es la CINÉTICA DE CADA UNA DE LAS MOLÉCULAS Y PARA UN MOL DE GAS SERÁ IGUAL A:

61
Energía interna de un gas ideal vs un gas real
El problema se complica cuando debemos calcularla para un gas real. En un gas real existen otras muchas formas de energía, y todas de acuerdo al enunciado forman parte de la ENERGÍA INTERNA, luego es lógico pensar en la dificultad de calcularlas una a una y posteriormente sumarlas: NO ES POSIBLE CALCULARLA

62
El calor en las transformaciones isobáricas ENTALPÍA
Una transformación isobárica es aquella que se verifica a P = cte. Podemos escribir entonces si sólo hay trabajo de expansión (o compresión): DE = qp + w = qp + (-p DV) = qp + [- p (Vf ‑ Vi)] Analicemos término por término: E es independiente del camino p es independiente del camino Vf es independiente del camino Vi es independiente del camino Luego DV será también independiente del camino DEBEMOS CONCLUIR QUE qp EN ESTE CASO SERÁ TAMBIÉN FUNCIÓN DE ESTADO (CUIDADO NO GENERALICE YA QUE ES UN EJEMPLO PARTICULAR)
: DE = qp + w = qp + (-p DV) = qp + [- p (Vf ‑ Vi)] Analicemos término por término: E es independiente del camino. p es independiente del camino. Vf es independiente del camino. Vi es independiente del camino. Luego DV será también independiente del camino. DEBEMOS CONCLUIR QUE qp EN ESTE CASO SERÁ TAMBIÉN FUNCIÓN DE ESTADO (CUIDADO NO GENERALICE YA QUE ES UN EJEMPLO PARTICULAR)")
63
ENTALPIA DE = qp + [-(P DV)] = qp + [-(P VB - P VA)]= EB ‑ EA
qp = (EB + P VB) ‑ (EA + P VA) Todos estos términos dependen del estado del sistema y no del camino. Al término "P.V" se lo llama Energía de volumen. E + P.V = CONTENIDO DE CALOR A PRESIÓN CONSTANTE, TAMBIEN LLAMADO ENTALPIA (H) H = E + PV
![ENTALPIA DE = qp + [-(P DV)] = qp + [-(P VB - P VA)]= EB ‑ EA](http://slideplayer.es/slide/5433484/17/images/63/ENTALPIA+DE+%3D+qp+%2B+%5B-%28P+DV%29%5D+%3D+qp+%2B+%5B-%28P+VB+-+P+VA%29%5D%3D+EB+%E2%80%91+EA.jpg "qp = (EB + P VB) ‑ (EA + P VA) Todos estos términos dependen del estado del sistema y no del camino. Al término P.V se lo llama Energía de volumen. E + P.V = CONTENIDO DE CALOR A PRESIÓN CONSTANTE, TAMBIEN LLAMADO ENTALPIA (H) H = E + PV.")
64
ENTALPIA LUEGO DEFINIMOS ENTALPIA (H) COMO LA SUMA DE LA ENERGÍA INTERNA MÁS LA ENERGÍA DE VOLUMEN EN UN INSTANTE DEFINIDO (RECUERDE QUE LOS TRES TÉRMINOS NO DEPENDEN DEL CAMINO) DH = DE + [- P. DV] y qp = HB ‑ HA = DH
 COMO LA SUMA DE LA ENERGÍA INTERNA MÁS LA ENERGÍA DE VOLUMEN EN UN INSTANTE DEFINIDO (RECUERDE QUE LOS TRES TÉRMINOS NO DEPENDEN DEL CAMINO) DH = DE + [- P. DV] y. qp = HB ‑ HA = DH.")
65
Transformaciones isócoras
Cuando DV = 0 (cero) se encuentra que DE = qV y por lo tanto no depende del camino, siendo: DEV = DHV
 se encuentra que. DE = qV. y por lo tanto no depende del camino, siendo: DEV = DHV.")
66
(J/ºC) o (J/K) (o cal en lugar de J)
Capacidad calorífica Definimos la Capacidad calorífica como la cantidad de calor necesario para elevar en un grado la temperatura de un sistema (J/ºC) o (J/K) (o cal en lugar de J)
 o (J/K) (o cal en lugar de J)")
67
Capacidad calorífica Calor específico: calor necesario para elevar en un grado la temperatura de 1 g de sustancia. Calor molar: calor necesario para elevar en un grado la temperatura de 1 mol de sustancia.

68
Capacidades caloríficas (1)
La capacidad calorífica nos da información sobre la energía interna Estructura molecular. Capacidades Caloríficas en gases. Ecuación válida para cualquier proceso Proceso isócoro

69
Capacidades caloríficas (2). Gas Ideal
Relación entre Capacidades Caloríficas en gases ideales. Proceso isóbaro Ecuación válida para cualquier proceso

70
Capacidad calorífica Capacidad calorífica de los gases ideales
LOS Cp y los Cv SON CARACTERÍSTICOS DE LAS SUSTANCIAS EN CONDICIONES DE PRESIÓN o VOLUMEN y TEMPERATURA DEFINIDOS Capacidad calorífica de los gases ideales

71
Capacidades caloríficas en gases y grados de libertad (2)
GASES MONOATÓMICOS l=3 (traslación) GASES DIATÓMICOS l= 3(tras.)+2(rot.) Además pueden vibrar y añadir un grado más de libertad a temperaturas altas
 GASES DIATÓMICOS. l= 3(tras.)+2(rot.) Además pueden vibrar y añadir un grado más de libertad a temperaturas altas.")
72
Gases ideales vs gases reales La tabla muestra algunos valores de Cp y Cv expresados en cal/grado.mol y que fueron hallados a 25°C GAS Cp Cv Ar 4,97 2,98 He 4,97 2,98 H ,90 4,91 O , ,05 CO , ,92 SO , ,30

73
TERMOQUÍMICA ET = COMPONENTE TÉRMICA O ENERGÍA TÉRMICA
EQ = COMPONENTE QUÍMICA O ENERGÍA QUÍMICA ES= ENERGÍA DEL SISTEMA = ET + EQ

74
ENERGÍA TÉRMICA Y ENERGÍA QUÍMICA
qV = DE = ET + EQ qP = DH = HT + HQ

75
ESTADOS NORMALES POR CONVENIO INTERNACIONAL SE ESTABLECIERON LAS CONDICIONES FÍSICAS BAJA LAS CUALES SE DEFINEN LOS ESTADOS NORMALES O ESTÁNDAR: ESTADO NORMAL DE UN GAS (puro): 1 atm y 25ºC ESTADO NORMAL DE UN LIQUIDO (puro): ESTADO NORMAL DE UN SÓLIDO ( ** ): atm y 25ºC ( ** ) puro y forma cristalina más estable
: 1 atm y 25ºC. ESTADO NORMAL DE UN LIQUIDO (puro): ESTADO NORMAL DE UN SÓLIDO ( ** ): 1 atm y 25ºC ( ** ) puro y forma cristalina más estable.")
76
ENTALPÍA DE LAS SUSTANCIAS PURAS
Sabiendo que la entalpía de una sustancia no se puede calcular, el camino es establecer una CONVENCIÓN ARBITRARIA LA ENTALPIA DE UNA SUSTANCIA QUÍMICA ELEMENTAL EN SU ESTADO NORMAL O ESTÁNDAR VALE CERO. H0 = 0 EL EXPONENTE 0 SIGNIFICA QUE LOS ELEMENTOS SE ENCUENTRAN EN ÉL ESTADO ESTÁNDAR

77
ENTALPÍA DE LAS SUSTANCIAS PURAS
A 1 atm y 25ºC, el azufre rómbico (sólido), el cobre sólido, el bromo líquido, el O2 gaseoso, el H2 gaseoso y el Na sólido tienen ENTALPIA IGUAL A CERO (0) Pero el O (g), el H (g), el Br (g) NO TIENEN ENTALPÍA CERO (0)
, el cobre sólido, el bromo líquido, el O2 gaseoso, el H2 gaseoso y el Na sólido tienen ENTALPIA IGUAL A CERO (0) Pero el O (g), el H (g), el Br (g) NO TIENEN ENTALPÍA CERO (0)")
78
ENTALPÍA DE LAS SUSTANCIAS PURAS
¿Cuándo es válido ésta definición? a) Se trabaja a temperatura constante (que se elige arbitrariamente, como por ej. 25º) b) Si hay calentamiento o enfriamiento se debe elegir otra temperatura a la cual H°=0.
 Se trabaja a temperatura constante (que se elige arbitrariamente, como por ej. 25º) b) Si hay calentamiento o enfriamiento se debe elegir otra temperatura a la cual H°=0.")
79
ENTALPÍA DE LAS SUSTANCIAS PURAS
c) Sólo las entalpías de los elementos (no de los compuestos) en sus estados más estables es cero. Ej.H(g) (hidrógeno monoatómico en estado gaseoso) no tiene entalpía igual a cero. Hg(s) tampoco tiene entalpía igual a 0 (ya que su forma más estable es la líquida) En los dos ejemplos las sustancias se encuentran a 25ºC y 1 atm.
 Sólo las entalpías de los elementos (no de los compuestos) en sus estados más estables es cero. Ej.H(g) (hidrógeno monoatómico en estado gaseoso) no tiene entalpía igual a cero. Hg(s) tampoco tiene entalpía igual a 0 (ya que su forma más estable es la líquida) En los dos ejemplos las sustancias se encuentran a 25ºC y 1 atm.")
80
ENTALPÍA DE LAS SUSTANCIAS PURAS
d) No se llevan a cabo transmutaciones nucleares ya que sino llegaríamos al absurdo de que una reacción como la que sigue no liberaría energía: 2 H2(g) He(g) donde ambas sustancias tienen de acuerdo con la convención indicada más arriba: H° = 0 Pero esta reacción libera una cantidad muy importante de energía: es la FUSIÓN NUCLEAR (reacción que produce energía en el sol)
 No se llevan a cabo transmutaciones nucleares ya que sino llegaríamos al absurdo de que una reacción como la que sigue no liberaría energía: 2 H2(g) He(g) donde ambas sustancias tienen de acuerdo con la convención indicada más arriba: H° = 0. Pero esta reacción libera una cantidad muy importante de energía: es la FUSIÓN NUCLEAR (reacción que produce energía en el sol)")
81
Entalpía de las reacciones

82
Entalpías de formación

83
Entalpías de formación

84
LEYES DE LA TERMOQUÍMICA
Primera Ley o Ley de Lavoisier-Laplace "EL CALOR NECESARIO PARA DESCOMPONER UNA SUSTANCIA EN SUS ELEMENTOS ES IGUAL AL CALOR LIBERADO CUANDO DICHA SUSTANCIA SE FORMA A PARTIR DE SUS ELEMENTOS"

85
LEYES DE LA TERMOQUÍMICA
H2(g) + ½ O2(g) H2O(g) DH°f= ‑241,60 kJ/mol De acuerdo a esta primera ley podemos escribir H2O(g) H2(g) + ½ O2(g) DH°-1= +241,60 kJ/mol
 + ½ O2(g) H2O(g) DH°f= ‑241,60 kJ/mol. De acuerdo a esta primera ley podemos escribir. H2O(g) H2(g) + ½ O2(g) DH°-1= +241,60 kJ/mol.")
86
LEYES DE LA TERMOQUÍMICA
Segunda ley de la Termoquímica o Ley de Hess En 1840 Hess postuló una ley absolutamente empírica: "el calor liberado a presión o volumen constante en una reacción química dada es una constante independientemente del número de etapas en que se realiza el proceso químico."

87
Segunda ley de la Termoquímica o Ley de Hess
C (s)(grafito) + ½ O2(g) CO(g) DH°r= ? Reacción difícil de lograr en el laboratorio: se aplica Hess (1) C(s)(grafito)+O2(g) CO2(g) DH°1=‑393,75 kJ/mol (2) CO(g) + ½ O2(g) CO2(g) DH°2=‑282,98 kJ/mol
(grafito) + ½ O2(g) CO(g) DH°r= Reacción difícil de lograr en el laboratorio: se aplica Hess. (1) C(s)(grafito)+O2(g) CO2(g) DH°1=‑393,75 kJ/mol. (2) CO(g) + ½ O2(g) CO2(g) DH°2=‑282,98 kJ/mol.")
88
Segunda ley de la Termoquímica o Ley de Hess
¿Qué podemos hacer con estas reacciones? En primer lugar apliquemos la primera ley a la reacción (2) que quedará: (‑2) CO2(g) CO(g) + ½ O2(g) DH°‑2= 282,98 kJ/mol De acuerdo a Hess podemos sumar la reacción (1) y la (‑2) (1) C(s)(grafito) + O2(g) CO2(g) (‑2)CO2(g) CO(g) + ½ O2(g) C(s)(grafito) + O2(g) + CO2(g) CO2(g)+CO(g)+½ O2(g) C(s)(grafito) + ½ O2(g) CO(g)
 que quedará: (‑2) CO2(g) CO(g) + ½ O2(g) DH°‑2= 282,98 kJ/mol. De acuerdo a Hess podemos sumar la reacción (1) y la (‑2) (1) C(s)(grafito) + O2(g) CO2(g) (‑2)CO2(g) CO(g) + ½ O2(g) C(s)(grafito) + O2(g) + CO2(g) CO2(g)+CO(g)+½ O2(g) C(s)(grafito) + ½ O2(g) CO(g)")
89
Segunda ley de la Termoquímica o Ley de Hess
¿Y el DH°r ? Para su cálculo se procede de idéntica manera que la realizada arriba con las ecuaciones: DH°1 + DH°‑2= ‑393,75+282,98 = ‑ 110,77 kJ/mol DH°r= ‑ 110,77 kJ/mol

90
Segunda ley de la Termoquímica o Ley de Hess
Lo mismo se puede graficar con un CICLO

91
Entalpías de combustión
C3H8(g) + 5O2(g) 3CO2(g) + 4H2O(l) El calor que se produce es de 2217,90 kJ/mol. Como se mide a 25ºC y 1 atm, ese valor es la ENTALPIA ESTÁNDAR DE COMBUSTIÓN: DH°c= ,90 kJ/mol
 + 5O2(g) 3CO2(g) + 4H2O(l) El calor que se produce es de 2217,90 kJ/mol. Como se mide a 25ºC y 1 atm, ese valor. es la ENTALPIA ESTÁNDAR DE COMBUSTIÓN: DH°c= -2217,90 kJ/mol.")
92
Equivalente Mecánico del Calor
1 caloría (energía calórica) = 4,183 Joules (enegía mecánica)
 = 4,183 Joules (enegía mecánica)")
Presentaciones similares








>")





 UAM 3.Termoquímica>")




