Descargar la presentación
La descarga está en progreso. Por favor, espere

1
Polineuroradiculopatía desmielinizante inflamatoria crónica en la infancia
Dr. Mariano De Sarratea Dra. Daniela Muñoz Residente 1°año INCA Neuróloga infantil

2
Introducción Enfermedad crónica inmunológicamente mediada del SNP
Debilidad muscular y disminución de ROT marcan el inicio de la enfermedad EMG , VCN y, en algunos casos Bx, demuestran proceso desmielinizante subyacente Historia natural altamente variable, la mayoría con buena recuperación european journal of p a e d i a t r i c neurology 1 6 ( ) e3 3 1
 e")
3
136 niños descritos en la literatura
Al igual que en adultos, Ig y corticoterapia primera opción de tratamiento Enfermedad relativamente rara en niños. Estudios retrospectivos con pequeños grupos de pctes y estudios de casos 136 niños descritos en la literatura european journal of p a e d i a t r i c neurology 1 6 ( ) e3 3 1
 e")
4
Epidemiología Prevalencia 1.0 - 1.9 x 100.000 hab
0,4 x en adultos entre años 0.73 x en > 55 años Mc leod et al: Incidencia 0.23 x en niños 0-9 años y 0.48 x entre años Prevalencia en niños: 0.5 x Presentación congénita descrita pero extremadamente rara european journal of p a e d i a t r i c neurology 1 6 ( ) e3 3 1
 e")
5
Patogenia: respuesta celular
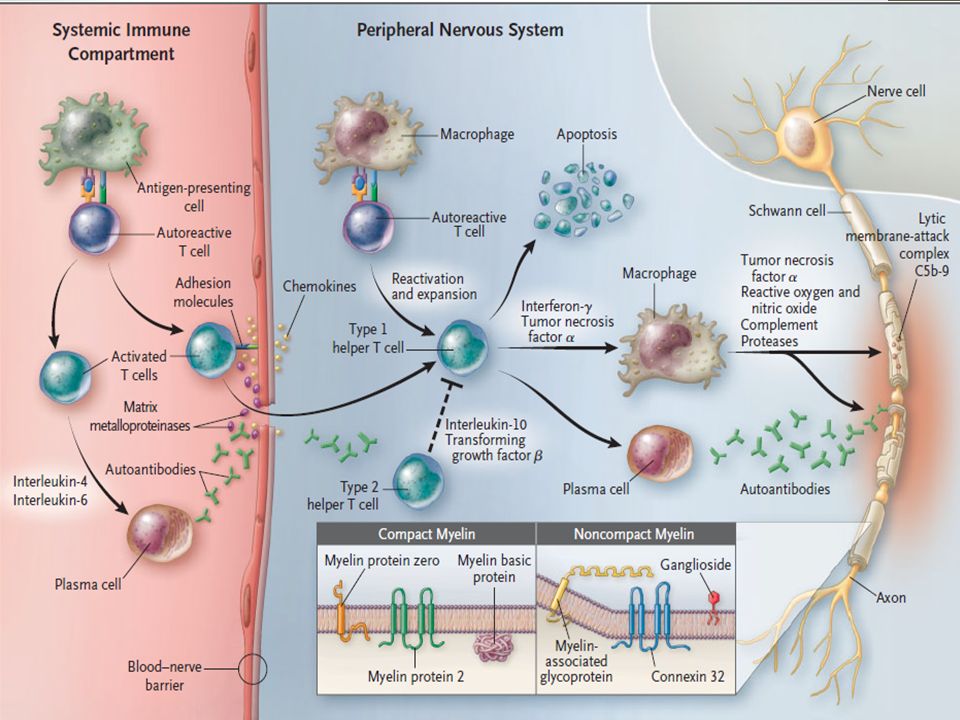
Linfocitos B y T activadas causan daño órgano especifico característico de las enfermedades autoinmunes Mimetismo molecular: El huésped genera una respuesta inmune en reacción a un organismo infeccioso el cual comparte epitopes con el tejido afectado (mielina) Sin embargo, en CIDP, sólo en contadas ocasiones se ha logrado determinar el target específico de esta respuesta
 Sin embargo, en CIDP, sólo en contadas ocasiones se ha logrado determinar el target específico de esta respuesta.")
6
Plasma y LCR: Niveles elevados de moléculas de adhesión, metaloproteinasas y citoquinas.
Downregulation de proteínas formadoras de tight-junction a nivel de la Membrana Basal (claudina-5 y ZO-1). Alteración de la barrera hemato-nerviosa Linfocitos T activados migran a través de la barrera hemato-nerviosa Ya en el SNP se produce expansión clonal de linfocitos T los cuales secretan TNF alfa, interferon gama e IL-2
. Alteración de la barrera hemato-nerviosa. Linfocitos T activados migran a través de la barrera hemato-nerviosa. Ya en el SNP se produce expansión clonal de linfocitos T los cuales secretan TNF alfa, interferon gama e IL-2.")
8
Reclutamiento de mas linfocitos y macrófagos los cuales fagocitan y generan acción citotóxica (radicales libres, metabolitos del oxido nítrico y proteasas) contra la mielina y las célula de schawnn Macrófagos también actuarían como presentadores de antígeno existiendo una sobreexpresión de complejo mayor de histocompatiblidad tipo 1 y 2

9
Patogenia: Respuesta Humoral
Deposito de Ig y complemento en fibras nerviosas mielinizadas Presencia de bandas oligoclonales de IgG en LCR Transferencia de IgG o suero de Pctes con CIDP induce bloqueos de condución y desmielinización en nervios de ratas a nivel experimental Gangliósidos y glicolípidos del nervio también pueden ser antígenos objetivo. Relación con infección por Campylobacter Jejuni.

10
Se comparten epítopes entre glicolípidos del nervio y lipopolisacáridos de la membrana microbiana.
Desmielinización y bloqueos de conducción también se pueden deber al ataque humoral mediante deposito de complemento Lo anteriormente expuesto lleva a una perdida axonal secundaria al proceso desmielinizante Pronóstico al largo plazo dependería mas bien de la perdida axonal que de la desmielinización Importancia de la terapia precoz

12
Clínica Mayoría con debilidad EEII, Alt. de la marcha y caídas frecuentes, dificultad para subir escaleras Debilidad de EESS puede estar presente, menos frecuente, puede ser el único síntoma Debilidad usualmente simétrica, aunque también ocasionalmente puede ser asimétrica Síntomas motores predominan en edad pediátrica, síntomas sensoriales menos frecuentes: parestesias y sensación de adormecimiento, ocasionalmente puede existir dolor de EE previo a la debilidad european journal of p a e d i a t r i c neurology 1 6 ( ) e3 3 1
 e")
13
Formas de presentación clínica
-Monofásica con recuperación completa (25% de los casos) -Recurrente remitente, la más frecuente -Lentamente progresiva -Progresión escalonada -Diagnóstico diferencial: Neuropatías inmunes agudas y neuropatías hereditarias motoras y sensitivas european journal of p a e d i a t r i c neurology 1 6 ( ) e3 3 1
 -Recurrente remitente, la más frecuente. -Lentamente progresiva. -Progresión escalonada. -Diagnóstico diferencial: Neuropatías inmunes agudas y neuropatías hereditarias motoras y sensitivas. european journal of p a e d i a t r i c neurology 1 6 ( ) e")
14
Alt. bulbares: Disfagia, hipofonía, fasciculaciones linguales
Distintas series han descrito menos comúnmente compromiso de pares craneano Síntomas oculomotores: Diplopía (+ frec), defecto pupilar aferente, paresia VI PC y ptosis Alt. bulbares: Disfagia, hipofonía, fasciculaciones linguales Debilidad facial generalizada, Alt. De la masticación, disgeusia y disartria también han sido descritos european journal of p a e d i a t r i c neurology 1 6 ( ) e3 3 1
, defecto pupilar aferente, paresia VI PC y ptosis. Alt. bulbares: Disfagia, hipofonía, fasciculaciones linguales. Debilidad facial generalizada, Alt. De la masticación, disgeusia y disartria también han sido descritos. european journal of p a e d i a t r i c neurology 1 6 ( ) e")
15
Atrofia muscular secundaria es frecuentemente observada
Compromiso pares craneanos puede en algunas ocasiones ser la única sintomatología Temblor y ataxia: Raro Atrofia muscular secundaria es frecuentemente observada Antecedente de infección previa 23 – 53%. Adultos 25 – 32% Síntomas respiratorios menos comunes que que en GBS: Ryan et al 3 de 16 pctes con Sx respiratorios leves Un caso excepcional descrito: Niña 11 años con parálisis unilateral de N. Frénico asociada evolucionó con falla respiratoria european journal of p a e d i a t r i c neurology 1 6 ( ) e3 3 1
 e")
16
Diagnóstico Criterios diagnósticos revisados durante el Grupo de trabajo de CIDP n°88 del Centro Europeo neuromuscular Basado en estos criterios: Dg Posible o Confirmado Presentación clínica variable, con Síntomas neuropáticos últimas 4 sem debidos a un proceso de desmielinización subyacente Caracterizado por desmielinización multifocal que afecta las raíces espinales, plexos y troncos nerviosos proximales o distales La confirmación se realiza con EMG, NCS y Bx Nervio sural european journal of p a e d i a t r i c neurology 1 6 ( ) e3 3 1
 e")
17
Criterios Clínicos -Debilidad muscular progresiva proximal o distal de EE superiores o inferiores de al menos 4 sem de evolución o, alternativamente, puede tener una progresión más rápida (presentación Guillian-Barré like) seguido de recurrencia o curso prolongado (más de 1 año) asociado a arreflexia o hiporreflexia Criterios de exclusión: -Historia o características clínicas de neuropatía hereditaria -Exposición a drogas o toxinas que causen neuropatía periférica -Hallazgos de laboratorio incluyendo Bx o estudios de ADN que muestran una etiología distinta de CIDP european journal of p a e d i a t r i c neurology 1 6 ( ) e3 3 1
 seguido de recurrencia o curso prolongado (más de 1 año) asociado a arreflexia o hiporreflexia. Criterios de exclusión: -Historia o características clínicas de neuropatía hereditaria. -Exposición a drogas o toxinas que causen neuropatía periférica. -Hallazgos de laboratorio incluyendo Bx o estudios de ADN que muestran una etiología distinta de CIDP. european journal of p a e d i a t r i c neurology 1 6 ( ) e")
18
Electromiografía Electrodiagnóstico de CIDP extensamente estudiado
En la practica el Dg esta basado en el hallazgo de un proceso de desmielinización asimétrica En ocasiones, debido al compromiso difuso y en parches el estudio electrofisiológico puede resultar negativo VCN en CIDP muestra enlentecimiento de las velocidades de conducción european journal of p a e d i a t r i c neurology 1 6 ( ) e3 3 1
 e")
20
El grado de enlentecimiento varía en diferentes partes de un nervio único y en partes equivalentes de nervios distintos en una misma extremidad La presencia de bloqueos de conducción y dispersión temporal son útiles en la detección de desmielinización segmental multifocal. Diferencia de patología hereditaria: ↓ VCN es uniforme a lo largo del nervio EMG y VCN deben realizarse idealmente en todos los casos sospechosos y ser realizados por un operador con experiencia con el objeto de aumentar las probabilidades de encontrar hallazgos compatibles con CIDP european journal of p a e d i a t r i c neurology 1 6 ( ) e3 3 1
 e")
21
Criterios Electrofisiológicos mayores:
-VCN menos de 75% del límite inferior normal en al menos 2 nervios -Latencias distales prolongadas en 2 o más nervios: > 130% del valor normal -Bloqueo de conducción y/o dispersión temporal del potencial de acción en al menos un nervio motor -Ausencia o prolongación de la latencia mínima de la onda F en dos o más nervios: > 130% del valor normal european journal of p a e d i a t r i c neurology 1 6 ( ) e3 3 1
 e")
22
Criterios electrofisiológicos de apoyo:
- Potenciales de acción sensoriales anormales de N. mediano con potencial de acción sensorial sural normal Indice de latencia mínima anormal Diferencia de más de 10 m/s en la VCN entre nervios de EESS e EEII o entre diferentes nervios de la misma extremidad european journal of p a e d i a t r i c neurology 1 6 ( ) e3 3 1
 e")
23
Dg electrofisiológico:
3 de 4 criterios mayores ó 2 criterios mayores más 2 criterios de apoyo

24
LCR Disociación albumino-citológica:
- Distintas series de casos, % Pctes. pediátricos tuvieron niveles > a 35 mg/dl -En algunos estudios pequeños el rango estuvo entre 41 y 305 mg/dl -En un caso llego a ser de 2150 mg/dl -En general el conteo celular es < a 10 cél/mm european journal of p a e d i a t r i c neurology 1 6 ( ) e3 3 1
 e")
25
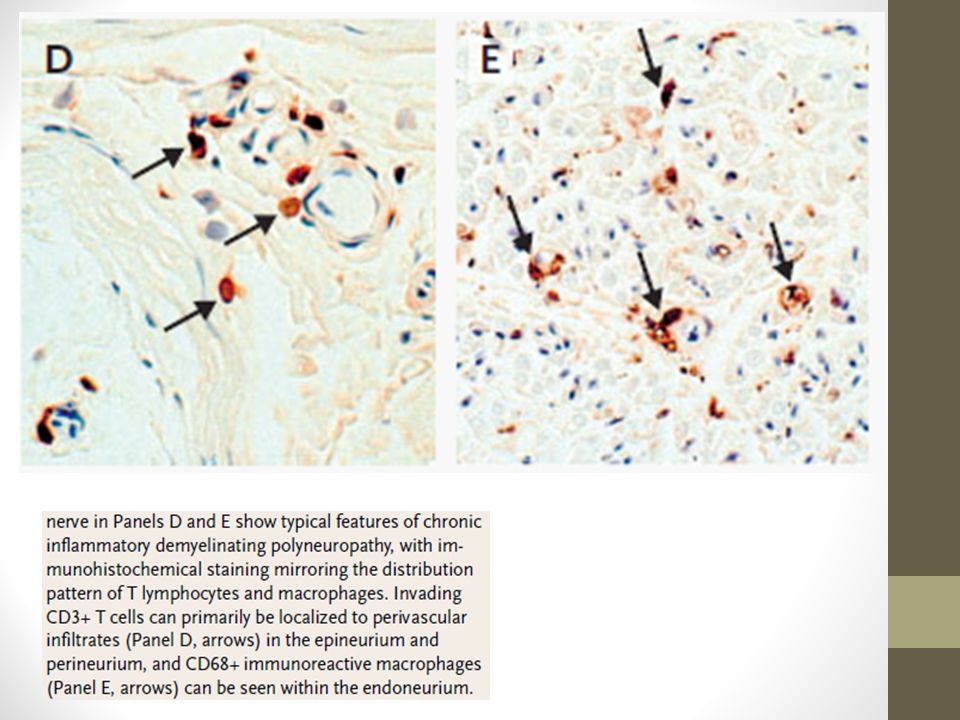
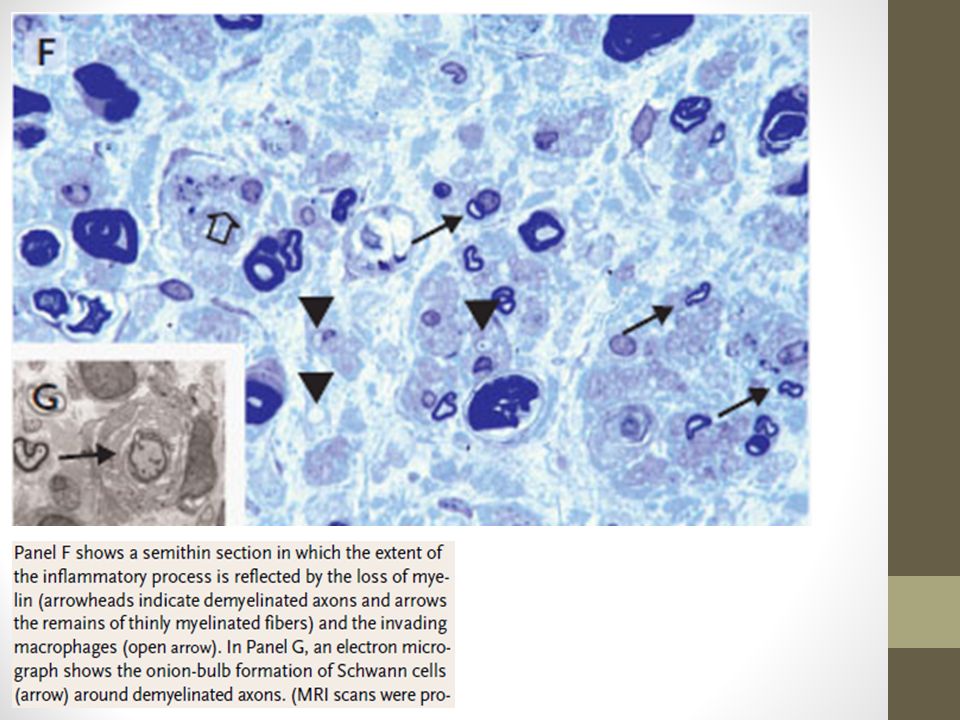
Biopsia No es imprescindible para diagnosticar CIDP. Resultado puede ser altamente variable La desmielinización es usualmente demostrada mediante la EMG y NCS. Sin embargo, la Bx puede apoyar el Dg cuando éste es dudoso Hallazgos pueden mostrar infiltración de mononucleares, desmielinización segmentaria, vaina de mielina adelgazada, remielinización y formación de bulbos de cebolla Edema del endoneuro y desmielinazción mediada por macrófagos también puede ser observada european journal of p a e d i a t r i c neurology 1 6 ( ) e3 3 1
 e")
27
RNM Hiperintensidad en T2 del plexo braquial o del nervio ciático
Refuerzo de raíces cervicales, del plexo braquial y plexo lumbar, realzan con la administración gadolinio

28
CIDP Confirmado Criterios clínicos + EMG + LCR
CIDP Posible Criterios clínicos + EMG o LCR o biopsia de nervio european journal of p a e d i a t r i c neurology 1 6 ( ) e3 3 1
 e")
30
Diagnóstico diferencial
ENF. CHARCOT-MARIE-TOOTH: Polineuropatía sensitivo-motora de tipo desmielinizante o axonal. Carácter hereditario. Frec 1 x 2500 NV, inicio Síntomas 1er y 2da década Tipos: 1 a, mutación PMP22 1 b, mutación P0 X, mutación conexina-32 Atrofia y debilidad muscular distal, asociada a perdida sensorial e hiporeflexia. Curso lento y progresivo Steppage, pie cavo y dedos en martillo, mano en garra, temblor y acrocianosis Arch Neurocien (Mex) INNN, 2012
 INNN,")
31
NEUROPATÍA HEREDITARIA CON PREDISPOSICIÓN A PARÁLISIS COMPRESIVA (NHPP):
Tendencia a desarrollar parálisis periférica ante mínimos traumatismos Tr. genético autosómico dominante deleción de la región 17p11 del gen PMP22 Formas más frecuentes de presentación son la polineuropatía sensitivo-motora desmielinizante así como también la mononeuropatía múltiple Más frec: N. mediano (canal del carpo), N. cubital en canal epitrocleo-olecraneano, N. peroneo (cabeza del peroné) Presentación en la Adolescencia o periodo adulto joven, recuperación clínica posterior a los episodios, curso recurrente Rev. chil. neuro-psiquiatr. vol.50 no.3 Santiago set. 2012
, N. cubital en canal epitrocleo-olecraneano, N. peroneo (cabeza del peroné) Presentación en la Adolescencia o periodo adulto joven, recuperación clínica posterior a los episodios, curso recurrente. Rev. chil. neuro-psiquiatr. vol.50 no.3 Santiago set")
32
DEFICIENCIA DE VITAMINA B12:
Desmielinización discontinua, difusa y progresiva de los cordones dorsales y laterales de la médula espinal Parestesias y perdida de la sensibilidad posicional y vibratoria, ataxia Puede haber disminución de ROT, hiperreflexia y espasticidad sobrevienen cuando se involucran los cordones laterales Distribución simétrica y distal, fundamentalmente en manos y pies Rev Cubana Hematol Inmunol Hemoter 1999;15(3):159-74
:")
33
INTOXICACIÓN POR ARSÉNICO:
Intoxicación crónica: Los síntomas incluyen cambios en la piel con hiperqueratosis, formación de verrugas en palmas y plantas de los pies Grandes áreas de hiperpigmentación intercalados entre pequeñas áreas de hipopigmentación en cara, cuello y espalda Daño neurológico muestra una neuropatía periférica simétrica con parestesia de extremidades distales, siendo las piernas mas afectadas que los brazos La neuropatía puede ser progresiva y con efectos dosis dependientes Biopsia: Axonopatía y desmielinización son los principales cambios en nervios periféricos Arch Neurocien (Mex) INNN, 2011
 INNN,")
34
Difteria, vasculitis, enfermedades sistémicas, DM, linfoma.
OTROS: Difteria, vasculitis, enfermedades sistémicas, DM, linfoma. Desórdenes neurometabólicos: Leucodistrofia metacromática, Enfermedad de Fabry, Neuropatía secundaria a Porfiria, Adrenoleucodistrofia european journal of p a e d i a t r i c neurology 1 6 ( ) e3 3 1
 e")
35
Tratamiento de primera línea
Corticoesteroides -Mejoría a corto plazo en 71 – 100% de la población pediátrica (en 1 a 4 semanas) -Dosis inicial: prednisona 1-2mg/Kg/día (máximo 60 – 80 mg/día), reducción de 5 mg cada 2 semanas. Pueden reaparecer los síntomas al intentar retirar los corticoides Ventajas: Menor costo, terapia oral menos invasiva -Opción Pulsos de metilprednisolona (menores RAMs) Efectos adversos: Osteopenia, retardo del crecimiento, cataratas, compromiso inmunitario, HTA, hiperglicemia, incremento de peso european journal of p a e d i a t r i c neurology 1 6 ( ) e3 3 1 europ ean j o u rnal of p a e d i a t r i c neurology 1 3 ( ) – 2 1 8
 -Dosis inicial: prednisona 1-2mg/Kg/día (máximo 60 – 80 mg/día), reducción de 5 mg cada 2 semanas. Pueden reaparecer los síntomas al intentar retirar los corticoides. Ventajas: Menor costo, terapia oral menos invasiva. -Opción Pulsos de metilprednisolona (menores RAMs) Efectos adversos: Osteopenia, retardo del crecimiento, cataratas, compromiso inmunitario, HTA, hiperglicemia, incremento de peso. european journal of p a e d i a t r i c neurology 1 6 ( ) e europ ean j o u rnal of p a e d i a t r i c neurology 1 3 ( ) –")
36
-Buena respuesta en 50-88% de CIDP pediátrico
Inmunoglobulina IV -Buena respuesta en 50-88% de CIDP pediátrico -Mejoría en 2 a 12 semanas -Puede utilizarse como primera opción , escasos efectos adversos -Algunos requerirán tratamientos recurrentes (mensuales o anuales) Mecanismo de acción poco claro: -Inhibición de la producción de autoanticuerpos -Neutralización de autoanticuerpos patogénicos -Bloqueo funcional del receptor Fc de macrófagos Efectos adversos: Poco frecuentes en niños. Cefalea, nauseas y fiebre durante la infusión european journal of p a e d i a t r i c neurology 1 6 ( ) e3 3 1 europ ean j o u rnal of p a e d i a t r i c neurology 1 3 ( ) – 2 1 8
 Mecanismo de acción poco claro: -Inhibición de la producción de autoanticuerpos. -Neutralización de autoanticuerpos patogénicos. -Bloqueo funcional del receptor Fc de macrófagos. Efectos adversos: Poco frecuentes en niños. Cefalea, nauseas y fiebre durante la infusión. european journal of p a e d i a t r i c neurology 1 6 ( ) e europ ean j o u rnal of p a e d i a t r i c neurology 1 3 ( ) –")
37
-Buena mejoría a corto plazo en 50 a 100%
Plasmaféresis -Buena mejoría a corto plazo en 50 a 100% -Reportes en niños con CIDP, esquemas: 3 sesiones de 6 días cada una 5 recambios de plasma cada dias Requiere repetir procedimiento -Eficacia dura desde 7 a 14 días (hasta 8 semanas) Beneficio corto plazo, logísticamente complejo debido a punciones venosas repetidas, no como primera opción en niños european journal of p a e d i a t r i c neurology 1 6 ( ) e3 3 1 europ ean j o u rnal of p a e d i a t r i c neurology 1 3 ( ) – 2 1 8
 Beneficio corto plazo, logísticamente complejo debido a punciones venosas repetidas, no como primera opción en niños. european journal of p a e d i a t r i c neurology 1 6 ( ) e europ ean j o u rnal of p a e d i a t r i c neurology 1 3 ( ) –")
38
Tratamiento segunda línea
Metotrexato Azatioprina Ciclosporina A Anticuerpos Monoclonales Interferon - Existe escasa experiencia en pacientes pediátricos europ ean j o u rnal of p a e d i a t r i c neurology 1 3 ( ) – 2 1 8
 –")
39
Pronóstico Remisión completa en 70 – 100% (adultos 65 – 70%)
Pacientes con progresión rápida: -Resolución completa >75% -Secuelas leves 25% Pacientes con progresión mayor a 3 meses: -Secuelas leves 78% -Secuelas severas 22% europ ean j o u rnal of p a e d i a t r i c neurology 1 3 ( ) – 2 1 8
 –")
40
Y.G.C ENF. CHARCOT-MARIE-TOOTH:
Sin Historia familiar, sin pie cavo ni dedos en martillo, no presenta mano en garra, sin temblor ni acrocianosis. No presentó curso lento y progresivo NEUROPATÍA HEREDITARIA CON PREDISPOSICIÓN A PARÁLISIS COMPRESIVA (NHPP): Sin traumatismos como gatillante, más frecuente en zonas susceptibles de atrapamiento
: Sin traumatismos como gatillante, más frecuente en zonas susceptibles de atrapamiento.")
41
Y.G.C. DEFICIENCIA DE VITAMINA B12:
Sin parestesias, sin perdida de la sensibilidad posicional, sin ataxia. Niveles Vit B12 normales INTOXICACIÓN POR ARSÉNICO: Sin antec de exposición. Sin alteraciones en la piel, sin parestesias distales en extremidades

42
Y.G.C. CIDP Confirmado cuadro cumple: criterios clinicos
electroFISIOLÓGICOS (emg y vcn) LCR CIDP Confirmado
 LCR. CIDP Confirmado.")
43
gGRACIAS

Presentaciones similares












 Estimulación ovárica.>")






>")

