Descargar la presentación
La descarga está en progreso. Por favor, espere

1
Enfermedades genéticas más prevalentes
Ismael Ejarque Doménech, MD, PhD Grupo de trabajo sobre “Genética Clínica y Enfermedades Raras” de la SEMFyC. Comisión de Atención Primaria de la Asociación Española de Genética Humana (AEGH). Jornada de Genética Clínica para Médicos de Familia Valencia, 2 de octubre de 2014
. Jornada de Genética Clínica para Médicos de Familia. Valencia, 2 de octubre de")
2
Introducción El “Royal College of General Practitioners” del Reino Unido estableció un Currículum de conocimientos en Genética Clínica que todo Médico de Familia británico debería conocer. Allí se establecen las enfermedades más frecuentes que se deben conocer. El Currículum formativo español del Médico de Familia NO aporta un apartado específico para habilidades en Genética Clínica.

7
Objetivo de habilidades en genética clínica para la AP
1) Identificación de personas a riesgo para una patología genética. 2) Asesoramiento preconcepcional. 3) Técnicas de diagnóstico prenatal. 4) Teratología. 5) Seguimiento clínico de los enfermos con enfermedades genéticas. 6) Identificación de problemas psicosociales. 7) Tests genéticos. 8) Recursos de Genética Clínica en internet. 9) Conocer los servicios de Genética más próximos, y como remitirlos. 10) Conocer las propias limitaciones.
 Identificación de personas a riesgo para una patología genética. 2) Asesoramiento preconcepcional. 3) Técnicas de diagnóstico prenatal. 4) Teratología. 5) Seguimiento clínico de los enfermos con enfermedades genéticas. 6) Identificación de problemas psicosociales. 7) Tests genéticos. 8) Recursos de Genética Clínica en internet. 9) Conocer los servicios de Genética más próximos, y como remitirlos. 10) Conocer las propias limitaciones.")
8
Paradigmas (I) Antes: …”quédese embarazada y luego la derivaremos a la unidad de genética…”
 Antes: … quédese embarazada y luego la derivaremos a la unidad de genética…")
9
Paradigmas (II) Ahora:
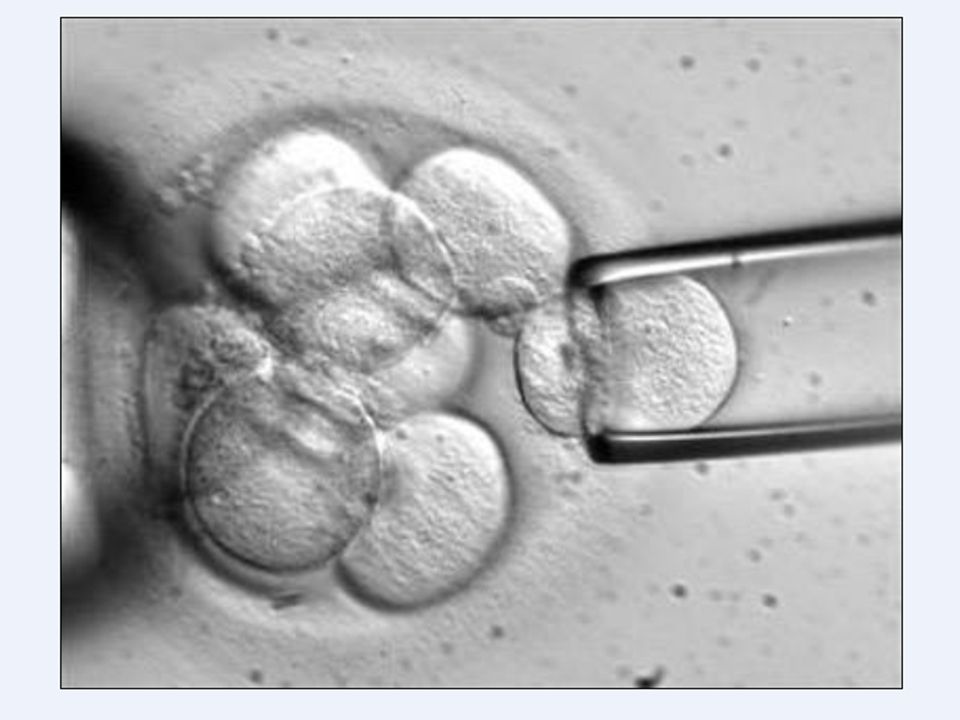
“Identificar a la pareja con riesgo de transmitir una enfermedad hereditaria ANTES DE la concepción y así derivarla a la unidad de genética de zona.” “ Diagnóstico Genético Preimplantación” (DGP).
.")
11
Ejemplos de anomalías cromosómicas frecuentes
Síndrome de Down. Síndrome de Turner. Síndrome de Klinefelter.

12
Ejemplos de enfermedades monogénicas: A.Dominantes
Neurofibromatosis. Enfermedad de Huntington. Hipercolesterolemia familiar.

13
Ejemplos de enfermedades monogénicas: A.Recesivas
Fibrosis quística. Hemoglobinopatías (enfermedad de células falciformes, talasemias). Hemocromatosis.
. Hemocromatosis.")
14
Ejemplos de enfermedades monogénicas: ligadas a X
Distrofias musculares de Duchenne y Becker. Hemofilia A y B. Síndrome del cromosoma X frágil.

15
Síndrome del X frágil Causa de retraso mental hereditario más frecuente. Debe su nombre a la rotura del cromosoma X en cultivos celulares en estos pacientes. Origen: patología del gen FMR1 (localizado en Xq27.3). Expansión del triplete CGG repetido. Herencia ligada al cromosoma X: ligada al sexo. Un gen (FMR1), tres patologías: Discapacidad intelectual. Menopausia precoz (POF). Ataxia en el adulto (FXTAS). Fenómeno de anticipación.
. Expansión del triplete CGG repetido. Herencia ligada al cromosoma X: ligada al sexo. Un gen (FMR1), tres patologías: Discapacidad intelectual. Menopausia precoz (POF). Ataxia en el adulto (FXTAS). Fenómeno de anticipación.")
16
I II III Menopausia precoz (<40) 90 Retraso mental
65 49 III 36 33

17
Del ADN a la proteína Promotor

18
Gen FMR1 Alelo normal : 5 – 50 (estable)
Proteina FMRP CGG CGG CGG CGG CGG CGG CGG FMR1 PROM Alelo normal : 5 – 50 (estable) Proteina FMRP CGG CGG CGG CGG CGG CGG CGG CGG CGG CGG CGG CGG CGG FMR1 PROM Alelo premutado : (inestable) CGG CGG CGG CGG CGG CGG CGG CGG CGG CGG CGG CGG CGG CGG CGG CGG CGG CGG FMR1 PROM Metilación Mutación completa: > 200 ¡Sin proteína FMRP!
 Proteina. FMRP. CGG CGG CGG CGG CGG CGG CGG CGG CGG CGG CGG CGG CGG. FMR1. PROM. Alelo premutado : (inestable) CGG CGG CGG CGG CGG CGG CGG CGG CGG CGG CGG CGG CGG CGG CGG CGG CGG CGG. FMR1. PROM. Metilación. Mutación completa: > 200. ¡Sin proteína. FMRP!")
19
Genotipo - Fenotipo: Repeticiones CGG
* Alelo normal (5-50). * Alelo premutado (50-200): - No Retraso Mental. - Riesgo de FXTAS (ataxia del adulto). - Riesgo de POF (menopausia precoz). * Mutación completa (>200): - Hombres: 100% Retraso Mental. - Mujeres: 50% Retraso Mental. 50% Normales.
. * Alelo premutado (50-200): - No Retraso Mental. - Riesgo de FXTAS (ataxia del adulto). - Riesgo de POF (menopausia precoz). * Mutación completa (>200): - Hombres: 100% Retraso Mental. - Mujeres: 50% Retraso Mental. 50% Normales.")
20
NINGÚN DATO ES PATOGNOMÓNICO
- Cara alargada. - Frente prominente. - Orejas grandes. - Mandíbula prominente. - Macroorquidismo. - Temperamento anormal. - Hiperactividad. - Ocasionalmente autismo, etc. NINGÚN DATO ES PATOGNOMÓNICO

21
FXTAS Sindrome de tremor/ataxia asociado a X-frágil.
Premutación en hombres: Ataxia cerebelar progresiva + temblor de intención. Otros hallazgos neurológicos: pérdida memoria corto plazo, declive cognitivo, parkinsonismo, etc. Penetrancia: dependiente de la edad. Premutación en mujeres: algunas desarrollan FXTAS. POF: Fallo ovárico precoz (menopausia precoz).
.")
22
FMR1-related POF Menopausia precoz.
POF: Fallo ovárico prematuro (< 40 años). Asociada a mujeres con premutación FMR1 CGG: repeticiones. Pero… ¡No toda menopausia precoz es debida a FMR1! Menopausia precoz.
. Asociada a mujeres con premutación FMR1. CGG: repeticiones. Pero… ¡No toda menopausia precoz es debida a FMR1! Menopausia precoz.")
23
Fenómeno de anticipación
Produce afectación cada vez más grave en las generaciones subsiguientes de una familia. El mecanismo subyacente es el incremento del número de repeticiones de tripletes en el gen FMR1 a través de las generaciones.

24
Síndrome de X frágil Signos guía de partida:
Criterios de derivación para test genético a través de los datos del árbol genealógico Signos guía de partida: 1. Discapacidad intelectual en hombre o mujer. 2. Menopausia precoz. 3. Alteración de la marcha / parkinsonismo.

25
? ? I II III Menopausia precoz (<40) 90 ¿Alelo premutado?
Retraso mental Menopausia precoz (<40) ¿Alelo premutado? ¿Mutación completa? 65 49 ¿Normales? ¿Alelo premutado? No es mutación completa. III ? ? 36 33
 ¿Alelo premutado ¿Mutación completa ¿Normales ¿Alelo premutado No es mutación completa. III")
26
Prevención de Retraso Mental por X frágil
Técnicas de Diagnóstico Prenatal: Amniocentesis. Biopsia de vellosidades coriales. Diagnóstico Genético Preimplantación (DGP).
.")
28
Ejemplos de anomalías cromosómicas frecuentes
Síndrome de Down. Síndrome de Turner. Síndrome de Klinefelter.

29
Síndrome de Down

30
Síndrome de Down Su incidencia aumenta con la edad materna.
Hay una gran incidencia de pérdidas fetales espontáneas con trisomía 21. La prevalencia de 1/600 por recién nacidos vivos ha disminuido a 1/1.100 debido al cribado prenatal, al diagnóstico prenatal y a la interrupción de una parte de los embarazos afectos.

31
Síndrome de Down Bases moleculares: Trisomía 21. Translocación.
Mosaico.

32
Síndrome de Down El riesgo de recurrencia de trisomía 21 se afecta por la edad materna y el mosaicismo germinal. Globalmente se asesora un riesgo de recurrencia ligeramente menor de 1% para mujeres menores de 39 años. Si un individuo tiene trisomía 21, no hay riesgo aumentado para sus familiares de segundo y tercer grado.

33
Síndrome de Down Si una mujer con síndrome de Down se queda embarazada, el riesgo de dicha trisomía en la descendencia es del 50%. La fertilidad en varones con síndrome de Down es excepcionalmente rara.

34
Síndrome de Down A los padres no se les hace el cariotipo si el cariotipo del afecto muestra trisomía 21 ó si es mosaico. Sin embargo el cariotipo parental es esencial si el cariotipo del afecto muestra síndrome de Down mediante translocación.

35
Síndrome de Down Historia familiar de síndrome de Down:
Más de un familiar afecto en el mismo lado de la familia: aumenta la posibilidad de una translocación robertsoniana que implique al cromosoma 21 por lo que el cariotipo del paciente es esencial.

36
Síndrome de Down Cribado prenatal. El riesgo de síndrome de Down se aumenta si: El pliegue nucal está aumentado en la ecografía de la semana Niveles anormales del cribado sérico materno, ej: triple escrínin (AFP, β-hCG y uE3) en la semana ADN fetal en sangre materna: nueva tecnología.
 en la semana ADN fetal en sangre materna: nueva tecnología.")
37
Síndrome de Down Diagnóstico prenatal:
La prueba definitiva es posible mediante biopsia de vellosidades coriales en la semana ó mediante amniocentesis en la semana Ambas pruebas son invasivas y conllevan un pequeño riesgo de aborto en los días sucesivos a la prueba (1/100 y 1/200 respectivamente).
.")
38
Síndrome de Down Plan de consulta para atención primaria: Historia:
Dibujar el árbol genealógico. Preguntar sobre cualquier otro familiar afecto. Edad de la madre en el momento del nacimiento del síndrome de Down. ¿Hay algún otro miembro de la familia visto en un servicio de genética clínica? Exploración (del neonato).
.")
39
Síndrome de Down Plan de consulta para atención primaria: Historia.
Exploración (del neonato): Características faciales. Braquicefalia y fontanela posterior patente. Dermatoglifos: pliegue palmar único y “sandal gap” entre 1º y 2º dedos de pies. Hipotonía. Corazón: cuidadosa valoración clínica (Eco) por alta incidencia de cardiopatías congénitas. Parámetros de crecimiento: altura, peso, etc… con tablas específicas.
: Características faciales. Braquicefalia y fontanela posterior patente. Dermatoglifos: pliegue palmar único y sandal gap entre 1º y 2º dedos de pies. Hipotonía. Corazón: cuidadosa valoración clínica (Eco) por alta incidencia de cardiopatías congénitas. Parámetros de crecimiento: altura, peso, etc… con tablas específicas.")
40
Síndrome de Down Manejo inicial en atención primaria:
La sospecha de que un neonato que estás viendo por primera vez podría tener síndrome de Down requerirá una interconsulta con un pediatra. Para confirmar el diagnóstico, un análisis rápido de FISH para +21 y/o cariotipo (según centros) será llevado a cabo para confirmar o rechazar el diagnóstico.
 será llevado a cabo para confirmar o rechazar el diagnóstico.")
41
Síndrome de Down Manejo en curso en atención primaria:
Áreas clave a considerar anualmente: Valoración oftalmológica (son frecuentes las cataratas y los defectos refractivos). Valoración auditiva. Obesidad. Enfermedad periodontal. Celiaquía. Función tiroidea. Signos precoces de demencia. Ser conscientes de artritis, subluxación atlantoaxial, diabetes mellitus, leucemia, apnea obstructiva del sueño y convulsiones.
. Valoración auditiva. Obesidad. Enfermedad periodontal. Celiaquía. Función tiroidea. Signos precoces de demencia. Ser conscientes de artritis, subluxación atlantoaxial, diabetes mellitus, leucemia, apnea obstructiva del sueño y convulsiones.")
44
Síndrome de Turner

45
Síndrome de Turner Turner describió las características de este síndrome en 1938, y en 1959 se encontró que las niñas con síndrome de Turner tienen un cromosoma X ausente (cariotipo 45,X). Afecta a 1/2.500 recién nacidas vivas. La mayoría de las concepciones Turner se pierden como abortos espontáneos.
. Afecta a 1/2.500 recién nacidas vivas. La mayoría de las concepciones Turner se pierden como abortos espontáneos.")
46
Síndrome de Turner Características físicas:
Hay considerable variación fenotípica. En muchas chicas Turner sólo se da la estatura baja. El fenotipo puede ser suficientemente sutil por lo que el cariotipo se debe incluir rutinariamente en la evaluación de niñas con estatura baja porque de otro modo podría fallarse en el diagnóstico.

47
Síndrome de Turner Características físicas:
Estatura baja y disgenesia gonadal: talla media en adulto es 147 cm. Malformaciones cardiovasculares: 15%-50%, coartación de aorta y defecto septo ventricular. Edema de manos y pies: en recién nacida. Anomalías renales: riñón en herradura, otras anomalías estructurales y agenesia en 1/3.

48
Síndrome de Turner Características físicas:
Pubertad y desarrollo sexual: pobre crecimiento, mínimo desarrollo mamario. Algunas mujeres tienen reglas y esto es más probable en presencia de línea celular 46,XX (mosaico). Educación: inteligencia normal pero IQ es puntos más bajo que los hermanos/as. Un leve retraso del habla y del lenguaje. Dificultades en habilidades espaciales, perceptivas y visuales. La mayoría de ellas van a colegios convencionales.
. Educación: inteligencia normal pero IQ es puntos más bajo que los hermanos/as. Un leve retraso del habla y del lenguaje. Dificultades en habilidades espaciales, perceptivas y visuales. La mayoría de ellas van a colegios convencionales.")
49
Síndrome de Turner Características físicas:
Comportamiento: problemas de ajuste social son frecuentes. Empleo: la mayoría lleva una vida adulta independiente. Fertilidad y embarazo: la mayoría son infértiles. FIV con donante de ovocitos ha resultado con éxito. Riesgo en el embarazo del 2% para disección aórtica. Sordera: muy frecuente.

50
Síndrome de Turner Genética:
El riesgo de recurrencia tras el nacimiento de un bebé con 45,X es muy bajo (<1%).
.")
51
Síndrome de Turner Genética:
Riesgos de la descendencia: la fertilidad natural es rara. El diagnóstico prenatal debe ofrecerse por el riesgo aumentado para trisomía 21 y 45,X. Las mujeres que portan reordenamientos estructurales están en riesgo para un feto femenino afecto de manera similar o un problema más grave en un feto varón que puede no ser viable. Es posible la biopsia de vellosidades o la amniocentesis.

52
Síndrome de Turner Genética:
Diagnóstico prenatal: debería ofrecerse la biopsia de vellosidades a sem o la amniocentesis a sem. Eco para translucencia nucal en sem para evidenciar el hidrops.

53
Síndrome de Turner Plan de consulta para atención primaria:
Exploración: Altura. Cuello corto, ancho, “webbed neck”, nacimiento posterior del pelo bajo. Manos y pies: tumefacción/edema, metacarpianos/metatarsianos cortos, sobre todo el 4º. Tórax: pezones separados, tórax en armadura. Cardiovascular: pulsos femorales retrasados, soplos cardiacos, HTA. Nevus pigmentados. Desarrollo sexual secundario y genitales externos.

55
Síndrome de Klinefelter

56
Síndrome de Klinefelter
47,XXY tiene una prevalencia de 1/600 – 1/800 nacimientos de varones. Es más frecuente conforme aumenta la edad materna: 1/2.500 a 33 años. 1/300 a 43 años. Normalmente diagnosticados prenatalmente como hallazgo inesperado en amniocentesis o en la vida adulta durante estudios de infertilidad. La mayoría permanecen sin diagnosticar.

57
Síndrome de Klinefelter
Genética: Los padres de niños 47,XXY: no se cariotipan de rutina. El riesgo de recurrencia es bajo (<1%). Su descendencia: los varones 47,XXY son generalmente infértiles. Para los pocos que son fértiles o con uso de ICSI, puede haber un incremento del riesgo de aneuploidías y se debe ofrecer Diagnóstico Genético Preimplantación (DGP) o amniocentesis.
. Su descendencia: los varones 47,XXY son generalmente infértiles. Para los pocos que son fértiles o con uso de ICSI, puede haber un incremento del riesgo de aneuploidías y se debe ofrecer Diagnóstico Genético Preimplantación (DGP) o amniocentesis.")
58
Síndrome de Klinefelter
Diagnóstico prenatal: Se debe ofrecer a los padres, aunque hay un fino balance entre un riesgo de recurrencia muy pequeño y el riesgo de pérdida del embarazo asociado al procedimiento. Está indicado si un paciente con Klinefelter concibe un embarazo a causa del riesgo aumentado de aneuploidías.

59
Síndrome de Klinefelter
Plan de consulta para atención primaria: Historia: Hitos del desarrollo: el habla y el lenguaje puede estar ligeramente retrasados, pero en la mayoría de chicos el desarrollo está dentro del rango normal. Se ha notado con frecuencia una tendencia hacia el comportamiento pasivo y no asertivo.

60
Síndrome de Klinefelter
Plan de consulta para atención primaria: Historia: Escolarización: hay un modesto decremento en el IQ de puntos al compararlo con hermanos, pero el IQ varía ampliamente entre 67 y 133. 2/3 tendrán más problemas con la lectura y la ortografía que sus iguales 46,XY. En un estudio el 70% recibió apoyo educativo adicional. La mayoría de los chicos 47,XXY tienen trabajos de menor habilidad pero no aumenta el paro. La variación es amplia y algunos Klinefelter seguirán carreras profesionales.

61
Síndrome de Klinefelter
Plan de consulta para atención primaria: Historia. Fertilidad: los hombres 47,XXY son generalmente infértiles. De vez en cuando tienen algo de semen en sus testículos, incluso cuando no hay en el eyaculado.

62
Síndrome de Klinefelter
Plan de consulta para atención primaria: Exploración. Altura: los hombres 47,XXY tienden a ser altos, con una altura final de 186 cm (comparado con los 177 cm de 46,XY).
.")
63
Síndrome de Klinefelter
Plan de consulta para atención primaria: Exploración. Los niños entran en la pubertad de manera normal. A mitad de la pubertad los testículos empiezan a involucionar, y los chicos desarrollan hipogonadismo hipergonadotrópico con testosterona disminuida. Los testículos son pequeños en la vida adulta y los hombres con Klinefelter son infértiles, con excepciones esporádicas. No tienen más frecuencia de ser homosexuales. Con la excepción de los testes pequeños, tienen genitales normales y relaciones sexuales normales.

64
Síndrome de Klinefelter
Estudios analíticos: Análisis del semen: la mayoría serán azoospérmicos, y raramente el mosaicismo puede permitir la producción de esperma y la subsiguiente fertilidad.

65
Síndrome de Klinefelter
Impacto sobre el paciente y/o la familia. La presentación adulta generalmente confina el impacto del diagnóstico a problemas de fertilidad.

66
Síndrome de Klinefelter
Manejo en atención primaria. Asesorar a los padres sobre si su hijo muestra dificultades de desarrollo, de comportamiento o educativas. Podrían pedir la ayuda de un profesional, pues la intervención precoz puede mejorar el resultado.

67
Síndrome de Klinefelter
Manejo en atención primaria. Derivar a un endocrinólogo pediátrico alrededor de los 10 años de edad para monitorizar el crecimiento, la medida de testosterona, FSH y LH. Si los estudios sugieren un hipogonadismo hipergonadotropo se debería ofrecer la suplementación con testosterona. Esta suplementación (inyecciones i.m. o parches) mejoran la autoestima, el crecimiento de vello facial y la líbido. También protege frente a osteoporosis.
 mejoran la autoestima, el crecimiento de vello facial y la líbido. También protege frente a osteoporosis.")
68
Síndrome de Klinefelter
Manejo en atención primaria. Para el manejo de la infertilidad, derivar a un especialista de medicina reproductiva para considerar inseminación artificial con donante, o concepción asistida con FIV e ICSI. El esperma para ICSI puede ser obtenida del eyaculado o de biopsia testicular.

69
Síndrome de Klinefelter
Manejo en atención primaria. Algunos autores de la literatura científica mencionan un riesgo aumentado de cáncer en los varones 47,XXY. Un estudio con hombres con Klinefelter encontró que la incidencia de cáncer global no está aumentada y no se justifican medidas de rutina de cribado.

70
Síndrome de Klinefelter
Manejo en atención primaria. El cáncer de mama es más frecuente en hombres XXY que en XY (riesgo relativo: 20); sin embargo la edad media al diagnóstico fue de 72 años y el riesgo actual (aproximadamente del 3%) es mucho más bajo que el de las mujeres.
; sin embargo la edad media al diagnóstico fue de 72 años y el riesgo actual (aproximadamente del 3%) es mucho más bajo que el de las mujeres.")
72
Ejemplos de enfermedades monogénicas: A. Dominantes
Neurofibromatosis. Enfermedad de Huntington. Hipercolesterolemia familiar.

73
Neurofibromatosis NF1 Enfermedad de von Recklinghausen.
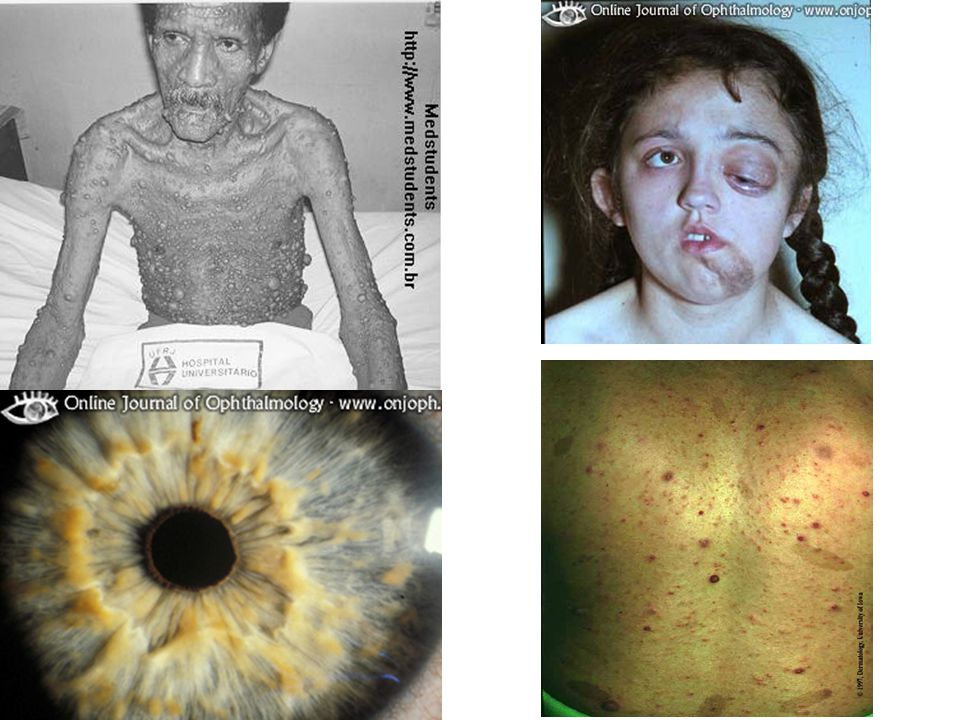
Autosómica Dominante: 17q11.2 1/2.500 al nacimiento: un médico de familia podría llegar a tener casi uno por cupo. NF1 vs NF2. NF2 más rara, sordera progresiva por schwannomas vestibulares: 22q.

75
Neurofibromatosis NF1 Criterios diagnósticos (dos o más):
Manchas café con leche: seis o más. Neurofibromas: dos o más. Pecas en axilas, cuello o ingles. Glioma óptico. Nódulos de Lisch en iris: dos o más. Lesión ósea: displasia del esfenoides, o adelgazamiento del córtex de huesos largos. Un familiar de primer grado con NF1.

76
Neurofibromatosis NF1 Genética (padres afectos):
La herencia es autosómica dominante con un 50% de riesgo para la descendencia. La tasa de nuevas mutaciones es del 50%.

77
Neurofibromatosis NF1 Genética (padres NO afectos):
Tras cuidadosa valoración clínica y estudio ocular. Una proporción pequeña pero significativa tiene mosaicos gonadales, con un riesgo de recurrencia más alto. Por tanto la derivación a un especialista y el asesoramiento son importantes.

78
Neurofibromatosis NF1 Genética:
NF1 es totalmente penetrante. Todas las mutaciones en NF1 dan manifestaciones clínicas. Muchas de las características muestran expresión dependiente de la edad. Como la NF1 muestra expresividad altamente variable, no es posible predecir las manifestaciones de la condición basado en la presencia o ausencia de complicaciones en otros miembros de la familia.

79
Neurofibromatosis Plan de consulta en medicina de familia. Historia:
Historia familiar detallada indagando específicamente en otros familiares con manchas café con leche, bultos o nódulos en su piel. Árbol genealógico de tres generaciones.

80
Neurofibromatosis Plan de consulta en medicina de familia.
Exploración. Piel: Las manchas café con leche que midan más de 0’5 cm que se incrementan con el paso del tiempo. Pecas en axilas, en nuca e ingles. El neurofibroma plexiforme desfigurante es aparente a partir de 2 años de edad. Los neurofibromas dérmicos aparecen desde la niñez tardía. Aumento generalizado de pigmentación cutánea, respecto a sanos.

82
Enfermedad de Huntington
Enfermedad neurológica progresiva con degeneración de los ganglios basales causando movimientos involuntarios, manifestaciones psiquiátricas y demencia. Causado por un número aumentado de repeticiones de tripletes CAG en el gen HD en 4p16. Se da fenómeno de anticipación por expansión de la repetición CAG.

83
Enfermedad de Huntington
Genética, herencia y riesgo de recurrencia: Autosómica dominante, con 50% de riesgo para los hijos/as. Factores que influencian la edad de inicio incluyen: edad de inicio de otros familiares y si la mutación se transmite por el padre o por la madre.

84
Enfermedad de Huntington
Historia: Edad de inicio de miembros afectos de la familia y edad de muerte. Enfermedad psiquiátrica, suicidio o demencia en miembros aparentemente no afectos (pueden haber sido EH). Árbol genealógico de 3 generaciones: extender más lejos si es posible para incluir todos los afectos conocidos.
. Árbol genealógico de 3 generaciones: extender más lejos si es posible para incluir todos los afectos conocidos.")
85
Enfermedad de Huntington
Manejo en Atención Primaria 1: Para un individuo sintomático que requiere derivación inicial para ayuda con el diagnóstico, derivar al Neurólogo, o en el caso de posible enfermedad juvenil a un Neuropediatra. Para aquellos más preocupados sobre su riesgo genético, o el de sus hijos/as, derivar a Genética Clínica.

86
Enfermedad de Huntington
Manejo en Atención Primaria 2: Esta condición tan lenta, de larga evolución y degenerativa necesitará un enlace cercano con todos los estamentos legales para asegurar la entrega de cuidados físicos y financieros apropiados. Cuando existan jóvenes madurando con un padre afecto con EH será necesario habilitar apoyo apropiado y consejo. Conocimiento del no-afecto, pero individuo “a riesgo” preocupado por su futuro.

88
Hipercolesterolemia Familiar
- Enfermedad hereditaria autosómica dominante. - Mutaciones en gen LDLR (19p13.2). - Codifica el receptor de las LDL. 5’ 3’ 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 PROMOTOR 1
. - Codifica el receptor de las LDL. 5’ 3’ PROMOTOR. 1.")
89
Clínica: · xantomas · xantelasma · arco corneal · CI precoz - Dos cuadros sindrómicos diferentes: · heterocigoto · homocigoto

90
Aproximadamente 1/500 de la población de Europa y Norteamérica son heterocigotos para mutaciones en LDLR. Hay mayor incidencia de HF en ciertas poblaciones como Afrikaaners, cristianos libaneses, fineses y franco-canadienses debido a efectos fundadores.

91
Hipercolesterolemia familiar
Historia: Árbol genealógico de tres generaciones con preguntas sobre familiares con hipercolesterolemia, infartos y angina. Historia de tenosinovitis del tendón de Aquiles.

92
Hipercolesterolemia familiar
Exploración: Explorar cuidadosamente xantomas tendinosos sobre los tendones aquíleos y sobre los tendones de los nudillos con los dedos extendidos. Comprobar la tensión arterial.

93
Hipercolesterolemia familiar
Estudios: Perfil lipídico en ayunas.

94
Hipercolesterolemia familiar
Manejo en Atención Primaria: Los pacientes afectos deberían estar bajo el cuidado de una clínica de lípidos y también por la Atención Primaria al mismo tiempo. Prescribir inicialmente estatinas y manejo a largo término con consejos sobre efectos adversos de los fármacos, monitorización anual del perfil lipídico... Consejos sobre estilo de vida saludable (tabaco, ejercicio, etc).
.")
96
Ejemplos de enfermedades monogénicas: A. Recesivas
Fibrosis quística. Hemoglobinopatías (enfermedad de células falciformes, talasemias). Hemocromatosis.
. Hemocromatosis.")
97
Fibrosis Quística

98
Fibrosis Quística FQ es la condición más frecuente autosómica recesiva en la población del norte de Europa. La incidencia en el norte de Europa es de 1/2.000 – 1/4.000 recién nacidos. Tasa de portadores sanos: 1/30. Parejas a riesgo: 1/30 x 1/30: 1/900 Al ser A. Recesiva: 1/4 x 1/900 = 1/3.600 Así, en teoría, uno de cada recién nacidos tendrá Fibrosis Quística.

99
Fibrosis Quística 3 fenotipos clínicos se asocian con mutaciones en CFTR (7q31-32): 1) FQ clásica: enfermedad pulmonar obstructiva, bronquiectasias, insuficiencia del páncreas exocrino, elevación del cloro en sudor e infertilidad en varones debido a ausencia bilateral congénita de conductos deferentes (CBAVD). Normalmente presente tras cribado neonatal, pero si no se detecta, en infancia hay fallo para medrar e infecciones pulmonares recurrentes.
 FQ clásica: enfermedad pulmonar obstructiva, bronquiectasias, insuficiencia del páncreas exocrino, elevación del cloro en sudor e infertilidad en varones debido a ausencia bilateral congénita de conductos deferentes (CBAVD). Normalmente presente tras cribado neonatal, pero si no se detecta, en infancia hay fallo para medrar e infecciones pulmonares recurrentes.")
100
Fibrosis Quística 2) FQ no-clásica: enfermedad pulmonar crónica ± enfermedad exocrina pancreática ± elevado cloro en sudor ± CBAVD. 3) Ausencia congénita bilateral de conductos deferentes (CBAVD).
 Ausencia congénita bilateral de conductos deferentes (CBAVD).")
101
Fibrosis Quística Genética, herencia y riesgo de recurrencia
Autosómica recesiva y por lo tanto el riesgo de recurrencia para los padres de un niño afecto es de ¼ ó el 25%.

102
Fibrosis Quística Plan de consulta en Atención Primaria. Historia.
Exploración. Enfermedad torácica crónica. Anomalías gastrointestinales y nutricionales. Síndromes de pérdida de sal. Azoospermia obstructiva.

103
Fibrosis Quística Exploración: Enfermedad torácica crónica:
Tos crónica y producción de esputos de colonización típica de patógenos FQ. Obstrucción al flujo aéreo (sibilantes). Sinusitis y obstrucción nasal por pólipos nasales. Anomalías gastrointestinales y nutricionales: Ileo meconial, prolapso rectal, obstrucción intestinal distal, insuficiencia pancreática, pancreatitis recurrente, fallo para medrar, hipoproteinemia y edema. Síndromes de pérdida de sales: Depleción aguda de sal y acidosis metabólica crónica. Azoospermia obstructiva (CBAVD).
. Sinusitis y obstrucción nasal por pólipos nasales. Anomalías gastrointestinales y nutricionales: Ileo meconial, prolapso rectal, obstrucción intestinal distal, insuficiencia pancreática, pancreatitis recurrente, fallo para medrar, hipoproteinemia y edema. Síndromes de pérdida de sales: Depleción aguda de sal y acidosis metabólica crónica. Azoospermia obstructiva (CBAVD).")
104
Fibrosis Quística Manejo inicial en Atención Primaria:
El médico de familia debería derivar, bajo sospecha, a un pediatra que colaborará con Genética Clínica, o directamente a Genética Clínica. El inicio de los síntomas suele ser en edad pediátrica.

106
Hemoglobinopatías Enfermedad de células falciformes. Talasemias.

107
Enfermedad células falciformes

108
Enfermedad células falciformes
En adultos normales el 97% de la hemoglobina es HbA (α2β2), entre 0-3’5% es A2 (α2γ2) y entre 0-1% es F (α2δ2). Sustitución de ácido glutámico por valina en 6ª posición de la cadena beta de globina HbS. Esto provoca que a menor presión de oxígeno, el eritrocito se deforme y adquiera apariencia de una hoz. La nueva forma provoca dificultad para la circulación de los eritrocitos. Por ello se obstruyen los vasos y causan síntomas como dolor en las extremidades. Los eritrocitos también tienen una vida media más corta anemia.
, entre 0-3’5% es A2 (α2γ2) y entre 0-1% es F (α2δ2). Sustitución de ácido glutámico por valina en 6ª posición de la cadena beta de globina HbS. Esto provoca que a menor presión de oxígeno, el eritrocito se deforme y adquiera apariencia de una hoz. La nueva forma provoca dificultad para la circulación de los eritrocitos. Por ello se obstruyen los vasos y causan síntomas como dolor en las extremidades. Los eritrocitos también tienen una vida media más corta anemia.")
109
Enfermedad células falciformes
Genética: Autosómica recesiva, cromosoma 11. Los individuos homocigotos recesivos sólo producen globina beta con valina. Afecta a 4/1.000 afroamericanos. Los individuos heterocigotos producen la mitad de la globina con ácido glutámico y la otra mitad con valina. Llevan vida bastante normal. Afecta a 8/1.000 afroamericanos. Ventaja para sobrevivir a malaria. A homocigotos y heterocitos, si planean un embarazo, se les puede ofrecer cribado de hemoglobinopatías. También Diagnóstico Genético Preimplantación (DGP). Antes del embarazo hay que derivar al servicio de Genética de zona.
. Antes del embarazo hay que derivar al servicio de Genética de zona.")
110
β-talasemias Conjunto de desórdenes sanguíneos hereditarios, caracterizado por anomalías en la síntesis de las cadenas beta de las hemoglobinas. Su consecuencia es un rango fenotípico que varía de la anemia severa a individuos clínicamente asintomáticos. La incidencia total anual de individuos sintomáticos es de 1 cada individuos en el mundo.

111
β-talasemias Las beta talasemias son causadas por mutaciones homocigotas puntuales, o, más raramente, por deleciones en el gen de las cadenas beta en el cromosoma 11, de la síntesis de dichas cadenas está reducida (beta+) o ausente (beta0). La transmisión es autosómica recesiva, sin embargo, mutantes dominantes han sido incluso notificadas. Producen anemia severa en homocigotos y heterocigotos compuestos.
 o ausente (beta0). La transmisión es autosómica recesiva, sin embargo, mutantes dominantes han sido incluso notificadas. Producen anemia severa en homocigotos y heterocigotos compuestos.")
112
β-talasemias Los heterocigotos para talasemia son el denominado rasgo talasémico. Suelen ser asintomáticos. Pueden tener anemia. A homocigotos y heterocitos, si planean un embarazo se les debería ofrecer cribado de hemoglobinopatías. También DGP. Antes del embarazo, derivar al servicio de genética de zona.

114
Hemocromatosis

115
Hemocromatosis Enfermedad del metabolismo del hierro que causa absorción excesiva de hierro de la dieta con su sobrecarga progresiva. Resulta en cirrosis hepática, diabetes mellitus, pigmentación cutánea y fallo testicular. Causado por mutaciones en el gen HFE del cromosoma 6.

116
Hemocromatosis En términos evolutivos los heterocigotos tenían una ventaja cuando las dietas eran pobres y había frecuentes infecciones por parásitos intestinales. En caucásicos 1/200 son homocigotos para la mutación C282Y. Pero esta mutación no es del todo penetrante y muchos de estos homocigotos nunca desarrollará clínica. La penetrancia se influencia por el sexo, la edad, el alcohol, otros factores genéticos y las pérdidas de hierro.

117
Hemocromatosis Genética: Hay dos mutaciones frecuentes: C282Y y H63D.
El 90% son homocigotos para C282Y. Herencia autosómica recesiva. La penetrancia es incompleta y la expresión es variable por lo que muchos que están predispuestos genéticamente nunca manifestará clínica seria.

118
Hemocromatosis Historia:
Obtener un árbol genealógico de 3 generaciones. Obtener una historia clínica detallada de los familiares afectos. Para los hijos buscar síntomas de hemocromatosis: Piel bronceada. Cirrosis hepática. Diabetes mellitus. Síntomas precoces: Debilidad y letargia. Artralgia. Impotencia o amenorrea. Disnea (cardiomiopatía).
.")
119
Hemocromatosis Exploración:
Manos por artropatía de pequeñas articulaciones. Pigmentación. Arritmias cardiacas. Signos de hipogonadismo.

120
Hemocromatosis Estudios en Atención Primaria: Metabolismo del hierro:
Ferritina: elevadas concentraciones (depósitos). Saturación de transferrina: > 50-55% indica acúmulo de hierro. Lo normal es 20-40%. Los portadores tienen niveles intermedios. ADN para HFE: tras informar al paciente. Si sintomático: función hepática, glucosa, AFP, LH, FSH (impotencia/amenorrea) y ECG.
. Saturación de transferrina: > 50-55% indica acúmulo de hierro. Lo normal es 20-40%. Los portadores tienen niveles intermedios. ADN para HFE: tras informar al paciente. Si sintomático: función hepática, glucosa, AFP, LH, FSH (impotencia/amenorrea) y ECG.")
121
Hemocromatosis Manejo en Atención Primaria:
Manejo de homocigotos y heterocigotos compuestos con metabolismo férrico anormal: Derivar al digestólogo. Cribado bioquímico de familiares de primer grado o derivar la familia al servicio de genética clínica. Riesgo mínimo de cirrosis hepática, pero si ferritina > 400/mg/L sangrías terapéuticas. /mg/L: donar sangre 4 veces al año. <200/mg/L: monitorizar para valorar acúmulo de hierro. Ferritina > mg/L Ferritina < mg/L

123
Ejemplos de enfermedades monogénicas: ligadas a X
Distrofias musculares de Duchenne y Becker. Hemofilia A y B. Síndrome del cromosoma X frágil.

124
Distrofias musculares

125
Distrofias musculares
Distrofia muscular de Duchenne: 1/3.000 – 1/4.000 varones recién nacidos. Forma más frecuente y grave de distrofia muscular en la niñez que acaba en muerte en los últimos años de la adolescencia o inicio de los 20 años. Es ligada a X recesiva por mutaciones en el gen de la distrofina (DMD) en Xp21. El gen DMD produce una proteína truncada.
 en Xp21. El gen DMD produce una proteína truncada.")
126
Distrofias musculares
Distrofia muscular de Becker: Parecida clínicamente a Duchenne pero más suave con edad media de inicio a 11 años y sobrevive hasta la media edad y más allá. Causado en el mismo gen que el de Duchenne, el DMD, pero en Becker las mutaciones producen proteína parcialmente y el fenotipo es más suave. Duchenne y Becker son ligadas a X.

127
Distrofias musculares
Formas de presentación: Los niños con Duchenne pueden presentar retraso de desarrollo, especialmente retraso del habla y andan tardíamente (>18 meses). Se pierde habilidad de andar independiente a 7-13 años y son dependientes de silla de ruedas. 30% de los niños tienen un leve discapacidad de aprendizaje que no es progresiva.
. Se pierde habilidad de andar independiente a 7-13 años y son dependientes de silla de ruedas. 30% de los niños tienen un leve discapacidad de aprendizaje que no es progresiva.")
128
Distrofias musculares
Formas de presentación: La cardiomiopatía es casi universal y la detección precoz puede permitir un tratamiento apropiado.

129
Distrofias musculares
Formas de presentación: En Becker la pérdida de la habilidad para caminar puede ocurrir tarde (ej: media edad o más tarde) con algunos pacientes permaneciendo ambulantes con ayudas. En adolescentes y década de 20 años la debilidad muscular es más evidente causando dificultad en caminar rápido, correr y subir escaleras Algunos pacientes presentan un fenotipo intermedio entre Duchenne y Becker.
 con algunos pacientes permaneciendo ambulantes con ayudas. En adolescentes y década de 20 años la debilidad muscular es más evidente causando dificultad en caminar rápido, correr y subir escaleras. Algunos pacientes presentan un fenotipo intermedio entre Duchenne y Becker.")
130
Distrofias musculares
Mujeres portadoras: Algunas pueden tener una elevación ligera de la creatinín kinasa (CK) pero otras lo tienen en el rango normal. Por tanto, una CK elevada indica portadora, pero una CK normal no lo excluye. Un pequeño porcentaje de mujeres son portadoras con clínica y tienen un grado variable de debilidad muscular.
 pero otras lo tienen en el rango normal. Por tanto, una CK elevada indica portadora, pero una CK normal no lo excluye. Un pequeño porcentaje de mujeres son portadoras con clínica y tienen un grado variable de debilidad muscular.")
131
Distrofias musculares
Genética, herencia y riesgo de recurrencia: Ligada a X recesiva: con 4 posibilidades de descendencia para una mujer portadora: varón normal, mujer normal, varón afecto y mujer portadora. La penetrancia es completa en los varones que heredan la mutación.

132
Distrofias musculares
Exploración: Observar la marcha del niño. Más evidente cuando tiene que darse prisa. Maniobra de Gower, donde el niño se flexiona sobre su muslo con sus manos para levantarse del suelo, debido a la debilidad muscular proximal. Raramente saltan con ambos pies juntos. Hipertrofia de pantorrillas en la mayoría.

133
Distrofias musculares
Estudios: Si se sospecha el diagnóstico derivar a neuropediatra o pediatra para valoración y estudios.

134
Distrofias musculares
Manejo en Atención Primaria Considerar vacunas de la gripe y la antineumococo. Distintivo de minusválido para el coche. Servicios sociales.

135
Distrofias musculares
Manejo de complicaciones a largo plazo: Pérdida de deambulación. Escoliosis. Hipoventilación nocturna. Problemas cardiacos. Discapacidad cognitiva.

137
Hemofilia A y B

138
Hemofilia A y B Sangrado debido a fallo en hemostasia secundaria. Defecto en estabilización del trombo. Factor VIII: hemofilia A, Xq28. Factor IX: hemofilia B, Xq27.

139
Genética Ligada a X recesiva: mujeres la portan y hombres la padecen.
Excepción: mujeres con inactivación desfavorable del X (lyonización) pueden tener hemofilia leve.
 pueden tener hemofilia leve.")
140
Presentación Sangrado espontáneo en las articulaciones y músculos o tras una lesión menor.

141
Historia Árbol genealógico de 3 generaciones con preguntas específicas: alteraciones del sangrado, sobre todo articular. También sangrado tras cirugía, extracción dental o amigdalectomía. Extender el árbol genealógico si se conocen otros varones afectos.

142
Exploración Derivar urgentemente si se sospecha hemartrosis articular.

143
Estudios Cuando el diagnóstico del probando es incierto, implicar al hematólogo. Hemograma: plaquetas. Cribado de la coagulación: tiempo de protrombina, tiempo de tromboplastina parcial activada. Un cribado normal no descarta Von Willebrand u otras enfermedades.

144
Manejo en Atención Primaria:
Complicaciones potenciales a largo plazo: Artropatía, con dolor, y pérdida de función. Muerte por sangrado (p.ej: hemorragia intracraneal). Infecciones transmitidas por transfusiones: VIH, VHB, VHC. Desarrollo de anticuerpos.
. Infecciones transmitidas por transfusiones: VIH, VHB, VHC. Desarrollo de anticuerpos.")
146
Test de compatibilidad genética
Antes de la concepción, a ambos miembros de la pareja. Estatus de portador sano para 600 enfermedades autosómicas recesivas. Si coindide el mismo gen para ser portador sano en ella y él: DGP o donante de gameto. Recombine.com IVI. Sanidad privada coste.

147
Teléfono Unidad de Genética del Hospital La Fe

148
ejarque_ism@gva.es www.gdtraras.es
Gracias por vuestra atención

Presentaciones similares