Descargar la presentación
La descarga está en progreso. Por favor, espere

1
INMUNODEFICIENCIAS Antonio González-Meneses López
Unidad de Dismorfología y síndrome de Down.

2
Objetivos Características del sistema inmune en el niño.
Formación Maduración Características específicas Alteraciones congénitas del sistema inmune. Inmunodeficiencias primarias. Alteraciones adquiridas del sistema inmune. Inmunodeficiencias secundarias.

3
Sistema inmune Pasivos: Activos (Sistema inmune): Piel Mucosas
Innata y adaptativa Linfocitos T, B y NK. Complemento. Fagocitos.

4
Sistema inmune activo Respuesta innata.
No mejora tras la exposición al antígeno. Complemento. Natural killers. Factores de coagulación Citoquinas Proteína C reactiva Respuesta adaptativa por memoria inmunológica. Mejora tras la exposición al antígeno. Linfocitos B Inmunoglobulinas. Linfocitos T Células dendríticas.

5
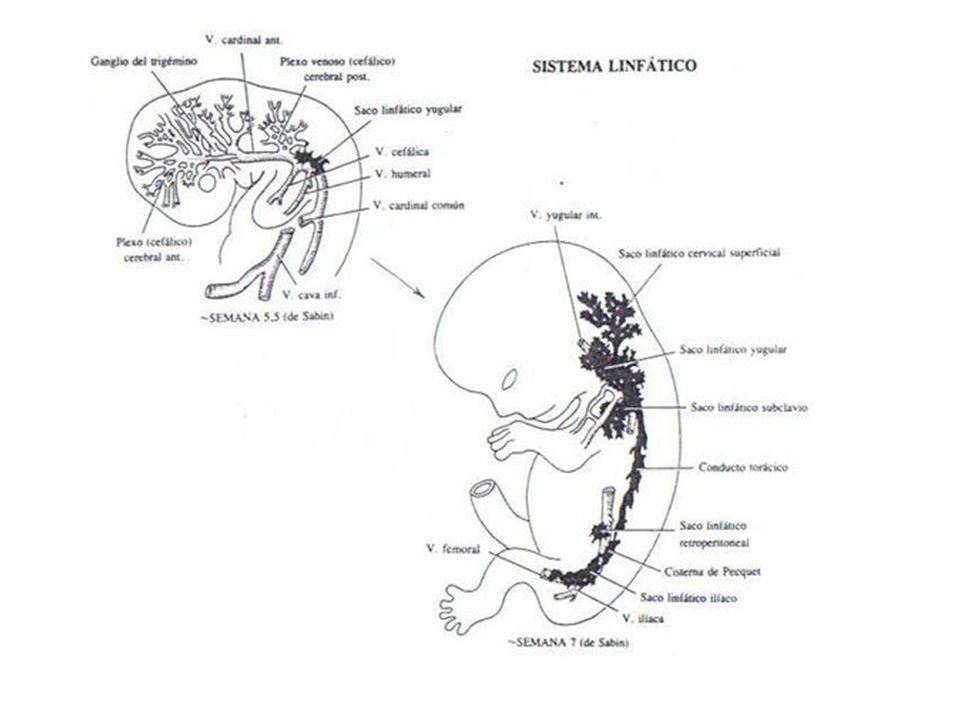
Génesis del sistema inmune
2-3 semanas, células madre hematopoyéticas pluripotenciales. 5 sem migran al hígado y médula ósea Diferenciación en linfocitos B, T y NK. Subtipos de estas son los linfocitos CD (clusters of differentiation). Linfocitos B y T: Reconocimiento específico de antígeno. Linfocitos NK: No reconocen un antígeno específico. Infecciones virales. Vigilancia tumoral. Citoquinas: Sustancias de relación entre las células defensivas.
. Linfocitos B y T: Reconocimiento específico de antígeno. Linfocitos NK: No reconocen un antígeno específico. Infecciones virales. Vigilancia tumoral. Citoquinas: Sustancias de relación entre las células defensivas.")
9
MADURACIÓN EMBRIONARIA DEL TIMO
Apoptosis de los linfocitos T que reaccionan frente a antígenos del organismo. Expresan CD4 y CD8. Migran del timo al bazo y ganglios en la semana y amigdalas en la 14. Maduros desde la semana 20 pero en bajo número.

10
Maduración linfocitos B
Migración al hígado con 7 semanas. Comienzan a expresar inmunoglobulinas precozmente (IgM). Maduran expresando IgD. A partir de la semana 14, reconocen antígenos. Ausentes hasta la semana 20, como células maduras.
. Maduran expresando IgD. A partir de la semana 14, reconocen antígenos. Ausentes hasta la semana 20, como células maduras.")
11
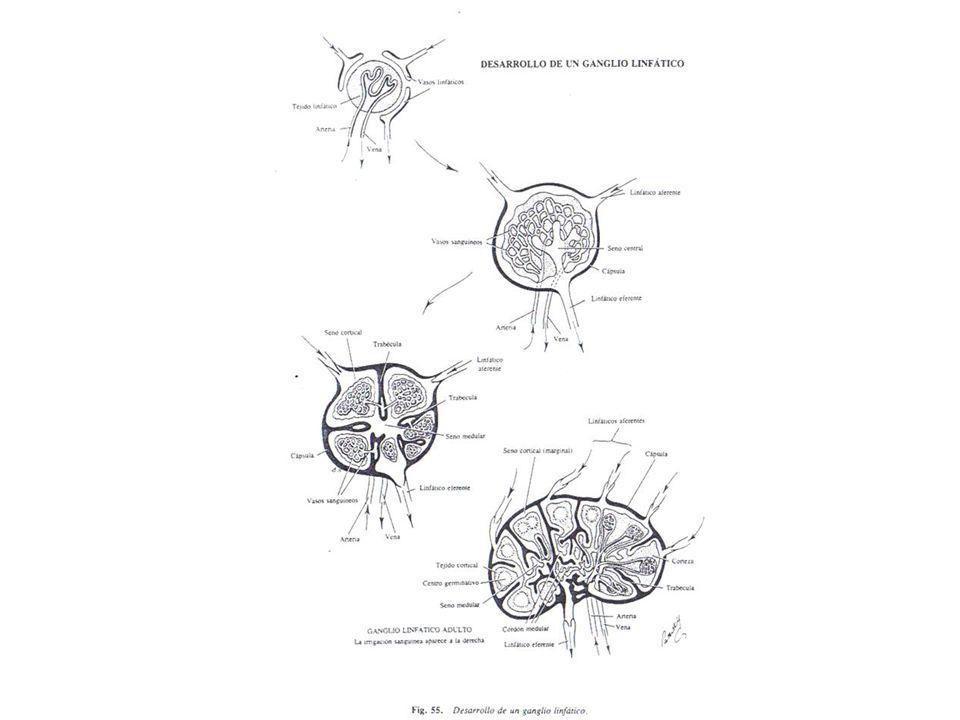
TIMO. GANGLIOS LINFATICOS. LINFOCITOS
TIMO. Su desarrollo precede ligeramente al de otros órganos linfáticos. GANGLIOS LINFATICOS Ø Al nacer: · Pequeños · Arquitectura folicular poco desarrollada · Ausencia de células plasmáticas Ø Durante 2-3 primeros meses: · Aumento tamaño · Incremento folículos linfoides, más ricos en linfocitos · Aparecen células plasmocitoides c. plasmáticas IMPACTOS ANTIGÉNICOS INMUNOGLOBULINAS PROPIAS LINFOCITOS T: Número circulante descendido. Madurez distintas variedades francamente disminuida.

12
Inmunoglobulinas IgM e IgG fijan complemento.
IgA: Secreciones externas. Fetal. Escasa al nacer. IgM: Fetal. Presente al nacer. IgG: PASA LA PLACENTA. Materna y fetal. Cantidades elevadas al nacer. IgD: Fetal. Escasa al nacer. IgE: IgM e IgG fijan complemento. IgG en todos los fluidos corporales.

14
Inmunoglobulinas La presencia de IgM elevada al nacer indica infección intraútero. Virus, protozoos. IgG fetales elevadas: Pueden ser maternas. Comparar con IgM. Diagnósticos diferenciales (VIH). Dificultan vacunas. Persistencia 6 meses.
. Dificultan vacunas. Persistencia 6 meses.")
15
Natural Killers Actividad citolítica .
No requieren memoria inmunológica. En el hígado desde la semana 11 desde la M. ósea. Posteriormente migran al bazo. Actúan contra microorganismos y células neoplásicas.

16
Complemento

17
RESUMEN SITUACION DEFENSIVA R.N.
MENOR CAPACIDAD LIBERARSE MICROORGANISMOS INFECTANTE. MAYOR PROPENSION A PADECER INFECCIONES. MAS FACILIDAD PARA GENERALIZACION PROCESO INFECCIOSO SEPSIS. MAYOR VULNERABILIDAD VARONES. El desarrollo de la inmunidad del niño se completa a la edad de 10 a 12 años

18
INMUNODEFICIENCIA Grupo de enfermedades de etiología diversa.
EN COMUN: fallo o defecto cuantitativo o cualitativo del sistema inmunitario ALTERADA DEFENSA CONTRA AGRESION EXTERNA EN CONSECUENCIA: INFECCIONES REPETIDAS NEOPLASIAS (fracaso defensa antitumoral) FENOMENOS AUTOINMUNES (falta reconocimiento antígenos propios) PATOLOGIAS ALERGICAS (respuesta anómala a antígenos ambientales)
 FENOMENOS AUTOINMUNES. (falta reconocimiento antígenos propios) PATOLOGIAS ALERGICAS (respuesta anómala a antígenos ambientales)")
19
Sospecha de inmunodeficiencia primaria
Ocho o más episodios de OMA al año Dos o más infec. graves de senos paranasales en un año. Dos o más meses de trat. antibiotico con poca mejoría Dos o más neumonías en un año Falta de ganancia de peso y talla. Infecciones recurrentes de piel y tejidos blandos y abscesos de órganos. Muguet o candidiasis cutánea persistente después del año de edad. Dos o más infecciones graves (sepsis, meningitis, osteomielitis). Necesidad de antibióticos intravenoso. Tipo de infección (proporciona una información valiosa sobre qué rama del sistema inmunitario está afectada), especialmente oportunista. Historia familiar de inmunodeficiencia.
. Necesidad de antibióticos intravenoso. Tipo de infección (proporciona una información valiosa sobre qué rama del sistema inmunitario está afectada), especialmente oportunista. Historia familiar de inmunodeficiencia.")
20
Inmunodeficiencias primarias
De la inmunidad específica. Síntesis de anticuerpos Déficit IgA. Agammaglobulinemia. Hiper IgM. Hipogammaglobulinemia transitoria lactante. Variable común. Combinadas. Combinada severa. Déficit de ADA. Déficit de PNP (purina nucleótido fosforilasa) Asociadas a defectos mayores Wiscot Aldrich. Di George. Ataxia telangiectasia. De la inmunidad natural o inespecífica. Déficits de complemento Deficiencia de factores. Angioedema hereditario. Deficiencias de fagocitosis Granulomatosis crónica. Deficiencia de la adhesión leucocitaria.
 Asociadas a defectos mayores. Wiscot Aldrich. Di George. Ataxia telangiectasia. De la inmunidad natural o inespecífica. Déficits de complemento. Deficiencia de factores. Angioedema hereditario. Deficiencias de fagocitosis. Granulomatosis crónica. Deficiencia de la adhesión leucocitaria.")
21
Inmunodeficiencias primarias (IP)
Habitualmente hereditarias (Ligadas a X o recesivas). Su sospecha puede salvar la vida del niño: conduce al diagnóstico y tratamiento (por ejemplo, con trasplante de progenitores hematopoyéticos). establecer medidas profilácticas frente a infecciones oportunistas . evitar actuaciones como la administración de vacunas de microorganismos vivos o sangre no irradiada. clínica: casi siempre, en primera infancia. puede niños mayores o incluso adultos, como: En la inmunodeficiencia variable común, los déficit aislados de IgA, algunas formas de agammaglobulinemia de Bruton, con mutaciones en el gen de la tirosincinasa de Bruton (Btk), que no dan lugar a una ausencia total de la proteína algunos tipos de enfermedad granulomatosa crónica.
. Su sospecha puede salvar la vida del niño: conduce al diagnóstico y tratamiento (por ejemplo, con trasplante de progenitores hematopoyéticos). establecer medidas profilácticas frente a infecciones oportunistas . evitar actuaciones como la administración de vacunas de microorganismos vivos o sangre no irradiada. clínica: casi siempre, en primera infancia. puede niños mayores o incluso adultos, como: En la inmunodeficiencia variable común, los déficit aislados de IgA, algunas formas de agammaglobulinemia de Bruton, con mutaciones en el gen de la tirosincinasa de Bruton (Btk), que no dan lugar a una ausencia total de la proteína. algunos tipos de enfermedad granulomatosa crónica.")
22
·INCIDENCIA 1/ / de los nacidos vivos, exceptuando la deficiencia aislada de la inmunoglubina A (IgA): Suele 1/700 (1/200-1/1.000 ) (mayoría detectada antes 15 años. 1º año 15% niños afectos) ·PREDOMINIO MASCULINO (h. ligada cromosoma X) 62% (80% <15 años). - Síntomas en varones. - Las mujeres portadoras sanas. ·PREDOMINIO POR DIAGNOSTICO: Inmunodeficiencias predominantemente humorales 50 al 68% " combinadas al 23% " celulares ,8 al 10,6% Deficiencia inespecífica ,5 al 22%
 ·PREDOMINIO MASCULINO (h. ligada cromosoma X) 62% (80% <15 años). - Síntomas en varones. - Las mujeres portadoras sanas. ·PREDOMINIO POR DIAGNOSTICO: Inmunodeficiencias predominantemente humorales 50 al 68% combinadas 14 al 23% celulares 6,8 al 10,6% Deficiencia inespecífica 7,5 al 22%")
23
Hipogammaglobulinemia con déficit de hormona de crecimiento
ETIOPATOGENIA MAYORIA ALTERACIONES GENETICAS CON TRANSMISION HEREDITARIA CONOCIDA (otras mutaciones "de novo") DESTACA. H. LIGADA AL CROMOSOMA X: Agammaglobulinemia Hipogammaglobulinemia con déficit de hormona de crecimiento Déficit de inmunoglobulinas con hiper IgM I. severa combinada S. de Wiskott-Aldrich
 DESTACA. H. LIGADA AL CROMOSOMA X: Agammaglobulinemia. Hipogammaglobulinemia con déficit de hormona de crecimiento. Déficit de inmunoglobulinas con hiper IgM. I. severa combinada. S. de Wiskott-Aldrich.")
24
AUTOSOMICA RECESIVA: Síndrome variable común
Deficiencia purina-necleotido-fosforilasa " de adenosina-deaminasa I. grave combinada ("síndrome del linfocito desnudo") Deficiencia de transcobalamina S. de ataxia-telangiectasia Deficiencia de complemento • AUTOSOMICO DOMINANTE: Déficit de inhibidor de C1
 Deficiencia de transcobalamina. S. de ataxia-telangiectasia. Deficiencia de complemento. • AUTOSOMICO DOMINANTE: Déficit de inhibidor de C1.")
25
·MALFORMATIVA ·DESCONOCIDA
S. Di George (Embriopatia o cromosomopatía: ausencia timo / aplasia paratiroide / faciales / cardiacas) Microdelección 22q11. Virus Epstein-Barr y Rubeola son teratogenos S.Inmune ·DESCONOCIDA
 Microdelección 22q11. Virus Epstein-Barr y Rubeola son teratogenos S.Inmune. ·DESCONOCIDA.")
26
Comienzo de la clínica Déficit de anticuerpos a partir 4-6 meses (perdida protección anticuerpos maternos). IP de células T: más grave y poco después del nacimiento (diarrea persistente, retraso de peso y talla e infecciones pulmonares) Defectos del sistema fagocítico poco después del nacimiento. No tomar en sentido estricto: Inmunodeficiencia variable común con deficiencia profunda de anticuerpos, aparece en cualquier época y, de hecho, es más frecuente en adultos Algunas mutaciones del gen de la Btk dan lugar a formas de agammaglobulinemia de Bruton que comienzan a manifestarse en niños mayores o en adultos.
 Defectos del sistema fagocítico poco después del nacimiento. No tomar en sentido estricto: Inmunodeficiencia variable común con deficiencia profunda de anticuerpos, aparece en cualquier época y, de hecho, es más frecuente en adultos. Algunas mutaciones del gen de la Btk dan lugar a formas de agammaglobulinemia de Bruton que comienzan a manifestarse en niños mayores o en adultos.")
27
Las inmunodeficiencias de células B
BACTERIANAS: Infecciones respiratorias (otitis media, sinusitis, bronquitis y neumonías) producidas por bacterias capsuladas como neumococo, Haemophilus influenzae, Staphylococcus aureus, Streptococcus pyogenes, Neisserias meningiditis y algunos gramnegativos como Pseudomonas aeruginosa (lesiones cutáneas induradas, negruzcas). Controladas con antibióticos, pero carácter recurrente (destrucción anatómica pulmón manifestada EPOC y bronquiectasias. VIRASICAS : Controlan la mayoría infecciones virales, pero son muy susceptibles a infecciones graves: meningoencefalitis crónica cuadro parecido a la dermatomiosistis, por enterovirus, sobre todo echovirus. Riesgo muy alto de enfermedad paralítica tras la vacunación con vacuna de poliovirus atenuados PROTOZOARIAS (lamblias).
 producidas por bacterias capsuladas como neumococo, Haemophilus influenzae, Staphylococcus aureus, Streptococcus pyogenes, Neisserias meningiditis y algunos gramnegativos como Pseudomonas aeruginosa (lesiones cutáneas induradas, negruzcas). Controladas con antibióticos, pero carácter recurrente (destrucción anatómica pulmón manifestada EPOC y bronquiectasias. VIRASICAS : Controlan la mayoría infecciones virales, pero son muy susceptibles a infecciones graves: meningoencefalitis crónica. cuadro parecido a la dermatomiosistis, por enterovirus, sobre todo echovirus. Riesgo muy alto de enfermedad paralítica tras la vacunación con vacuna de poliovirus atenuados. PROTOZOARIAS (lamblias).")
28
Déficit selectivo de IgA
La inmunodeficiencia congénita más frecuente. 1: nacidos vivos (1:700). Nivel sérico de IgA <5 mg/dL. Puede asociarse a déficit de IgG2. Puede faltar IgA secretora con IgA normal en suero. Infecciones respiratorias de predominio. Menos, neoplasias y autoinmunes. Frecuente la alergia o el asma alérgica. PUEDE CURSAR SIN INFECCIONES FRECUENTES. No tiene tratamiento específico.
. Nivel sérico de IgA <5 mg/dL. Puede asociarse a déficit de IgG2. Puede faltar IgA secretora con IgA normal en suero. Infecciones respiratorias de predominio. Menos, neoplasias y autoinmunes. Frecuente la alergia o el asma alérgica. PUEDE CURSAR SIN INFECCIONES FRECUENTES. No tiene tratamiento específico.")
29
Agammaglobulinemia de Bruton
Ligada al cromosoma X. Tirosina-cinasa de Bruton. Sin clínica mientras persiste IgG materna. Infecciones piogénicas extracelulares: Neumococo, Streptococo, Haemophilus Virasis bien toleradas salvo hepatitis y enterovirus. Parálisis con vacunas antipolio virus vivos.

30
Agammaglobulinemia de Bruton
Diagnóstico: Todas las inmunoglobulinas por debajo del 95%. Hipoplasia de amigdalas y ganglios linfáticos. Sin folículos ni centros germinales. Ausencia de respuesta inmune a las vacunas D dif con la hipogammaglobulinemia transitoria del lactante Función y número normal de los linfocitos T. Variante con talla baja y déficit de hormona de crecimiento. Tratamiento: Inmunoglobulinas IV periódicas. Antibióticos.

31
Inmunodeficiencia variable común
Similar al Bruton pero: Afecta a ambos sexos. Menor intensidad en la gravedad y comienzo más tardío. Ganglios linfáticos y amigdalas normales. Déficit sérico de todas las Inmunoglobulinas. Relación con el Déficit de IgA. (familiares o afectación secuencial). Cierta afectación inmunidad celular.
. Cierta afectación inmunidad celular.")
32
Hipogammaglobulinemia transitoria de la infancia (o del lactante).
Cuadro benigno. Niveles bajos de Inmunoglobulinas que se recuperan progresivamente. Se inmunizan con las vacunas. No requieren gammaglobulinas IV.

33
Déficit de subclases de IgG
IgG totales normales. Déficit de algunas subclases. IgG2 baja asociada a IgA baja. A veces progresan a un déficit total de Ig (IDVC). No deben recibir Ig IV de forma habitual.
. No deben recibir Ig IV de forma habitual.")
34
Enfermedad de Duncan (enfermedad linfoproliferativa ligada al X)
Insuficiente respuesta inmunitaria al virus Epstein- Barr. Ligada al X. Infección grave por el EBV con necrosis hepática extensa. Asocia déficit de inmunidad celular al EBV.

36
Síndrome hiper IgM IgM muy elevada con IgG e IgA bajas.
Considerada previamente déficit humoral. Hiperplasia linfoide asociada. Ligada al cromosoma X. Frecuentes alteraciones autoinmunes asociadas. Neutropenia cíclica frecuente. Alteración del CD40L. Interacciona con el CD40 y permite madurar al linfocito B.

37
Síndrome de Di George. Dismorfogénesis de la 3ª-4ª bolsa faríngea.
Hipoplasia tímica y de paratiroides. Déficit inmunitario celular. Hipocalcemia. Alteraciones cardiacas troncoconales. Retraso mental en grado variable. Delección 22q11.

39
Inmunodeficiencia combinada grave
Ausencia de función de las células T y B desde el nacimiento. Heterogenicidad genética. Muerte en el primer año de vida salvo trasplante de médula ósea precoz. Infecciones graves por gérmenes variados y oportunistas, especialmente diarreas, fallo de medro e infecciones respiratorias graves víricas o bacterianas. Linfopenia precoz. Inmunoglobulinas ausentes o disminuídas. Incapacidad para rechazar transplantes. Enfermedad de injerto contra huésped. Transfusiones irradiadas y desleucocitadas. Existen diferentes variedades.

40
Inmunodeficiencia combinada (Nezelof).
Infecciones graves y frecuentes: Infecciones pulmonares. Candiasis bucal o cutánea. Sepsis por gram negativos. Infecciones urinarias. Varicela grave. Parecido al SIDA. Niveles de inmunoglobulinas: Normales o elevados, con déficits ocasionales. Neutropenia y eosinofilia. Grave déficit de CD3 con CD4 y CD8 normales. Timo displásico. Ganglios linfáticos con deplección cortical y paracortical sin centros germinales.

41
Déficit de ADA (Adenosín deaminasa)
Autosómico recesiva. 15% de los pacientes con IDCG. Déficit inmunitario secundario a toxicidad linfocítica. Profundos déficits de todos los linfocitos. Lesiones óseas sugestivas de raquitismo. Terapia enzimática sustitutiva o TMO.

42
Wiskott-Aldrich Inmunodeficiencia con trombocitopenia y eccema.
Recesivo ligado a X. Hemorragias frecuentes secundarias a déficit plaquetario. Falta de respuesta a la inmunización. Wiskott-Aldrich

43
Ataxia-telangiectasia
Ataxia cerebelosa progresiva. Telangiectasias oculocutáneas. Inmunodeficiencia combinada variable. Susceptibilidad a tumores. Déficit de reparación del ADN al romperse. Autosómico recesiva.

Presentaciones similares












 ESPECÍFICAS (Respuesta inmunitaria) – La unión antígeno anticuerpo es específica.>")











