Descargar la presentación
La descarga está en progreso. Por favor, espere

1
TRASTORNOS NERVIOSOS Y MUCULARES

2
(G60-G64) Polineuropatías y otros trastornos del sistema nervioso periférico
(G60) Neuropatía hereditaria e idiopática (G60.0) Neuropatía hereditaria motora y sensorial (G60.1) Enfermedad de Refsum (G60.2) Neuropatía en asociación con ataxia hereditaria (G60.3) Neuropatía idiopática progresiva (G60.8) Otras neuropatías hereditarias e idiopáticas (G60.9) Neuropatía hereditaria e idiopática sin especificar (G61) Polineuropatía inflamatoria (G61.0) Síndrome de Guillain-Barré (G61.1) Neuropatía del serum (G61.8) Otras polineuropatías inflamatorias (G61.9) Polineuropatía inflamatoria sin especificar (G62) Otras polineuropatías (G62.0) Polineuropatía inducida por medicamentos (G62.1) Polineuropatía alcohólica (G62.2) Polineuropatía debida a otros agentes tóxicos (G62.8) Otras polineuropatías especificadas (G62.9) Polineuropatías sin especificar (G63) Polineuropatía en enfermedades clasificadas en otra parte (G64) Otros trastornos del sistema nervioso periférico
 Neuropatía hereditaria e idiopática. (G60.0) Neuropatía hereditaria motora y sensorial. (G60.1) Enfermedad de Refsum. (G60.2) Neuropatía en asociación con ataxia hereditaria. (G60.3) Neuropatía idiopática progresiva. (G60.8) Otras neuropatías hereditarias e idiopáticas. (G60.9) Neuropatía hereditaria e idiopática sin especificar. (G61) Polineuropatía inflamatoria. (G61.0) Síndrome de Guillain-Barré. (G61.1) Neuropatía del serum. (G61.8) Otras polineuropatías inflamatorias. (G61.9) Polineuropatía inflamatoria sin especificar. (G62) Otras polineuropatías. (G62.0) Polineuropatía inducida por medicamentos. (G62.1) Polineuropatía alcohólica. (G62.2) Polineuropatía debida a otros agentes tóxicos. (G62.8) Otras polineuropatías especificadas. (G62.9) Polineuropatías sin especificar. (G63) Polineuropatía en enfermedades clasificadas en otra parte. (G64) Otros trastornos del sistema nervioso periférico.")
3
(G70-G73) Enfermedades musculares y de la unión neuromuscular
(G70) Miastenia gravis y otros trastornos neuromusculares (G70.0) Miastenia gravis (G70.1) Trastornos mioneuronales tóxicos (G70.2) Miastenia congénita y del desarrollo (G71) Trastornos primarios de los músculos (G71.0) Distrofia muscular (G71.1) Trastornos miotónicos (G71.2) Miopatías congénitas (G71.3) Miopatía mitocondrial no clasificada en otra parte (G72) Otras miopatías (G72.0) Miopatía inducida por medicamentos (G72.1) Miopatía alcoholica (G72.2) Miopatía debida a otros agentes tóxicos (G72.3) Parálisis periódica
 Miastenia gravis y otros trastornos neuromusculares. (G70.0) Miastenia gravis. (G70.1) Trastornos mioneuronales tóxicos. (G70.2) Miastenia congénita y del desarrollo. (G71) Trastornos primarios de los músculos. (G71.0) Distrofia muscular. (G71.1) Trastornos miotónicos. (G71.2) Miopatías congénitas. (G71.3) Miopatía mitocondrial no clasificada en otra parte. (G72) Otras miopatías. (G72.0) Miopatía inducida por medicamentos. (G72.1) Miopatía alcoholica. (G72.2) Miopatía debida a otros agentes tóxicos. (G72.3) Parálisis periódica.")
4
(G73) Trastornos de la unión neuromuscular en enfermedades clasificadas en otra parte
(G73.0) Síndromes miasténicos en enfermedades endocrinas (G73.1) Síndrome de Eaton-Lambert (G73.2) Otros síndromes miasténicos en enfermedades neoplásticas (G73.3) Síndromes miasténicos en otras enfermedades clasificadas en otra parte (G73.4) Miopatía en enfermedades infecciosas y parásitas clasificadas en otra parte (G73.5) Miopatía en enfermedades endocrinas (G73.6) Miopatía en enfermedades metabólicas (G73.7) Miopatía en otras enfermedades clasificadas en otra parte
 Síndromes miasténicos en enfermedades endocrinas. (G73.1) Síndrome de Eaton-Lambert. (G73.2) Otros síndromes miasténicos en enfermedades neoplásticas. (G73.3) Síndromes miasténicos en otras enfermedades clasificadas en otra parte. (G73.4) Miopatía en enfermedades infecciosas y parásitas clasificadas en otra parte. (G73.5) Miopatía en enfermedades endocrinas. (G73.6) Miopatía en enfermedades metabólicas. (G73.7) Miopatía en otras enfermedades clasificadas en otra parte.")
5
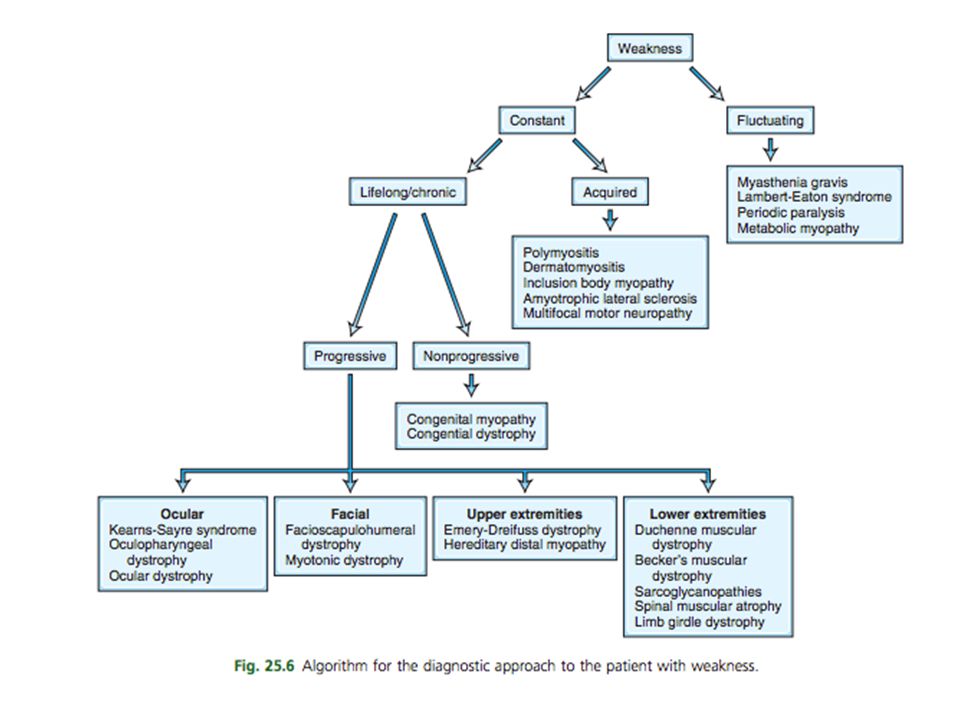
CONCEPTOS Neuropatía periférica: trastorno de los nervios periféricos sea cual sea su causa Se clasifican en simétricas y asimétricas (según la distribución de la afectación) y en axónicas, desmielinizantes o neuronales Polineuropatías: procesos de instauración gradual, que afectan a múltiples troncos nerviosos y que se caracterizan por ser simétricos y generalizados, con afectación preferentemente distal
 y en axónicas, desmielinizantes o neuronales. Polineuropatías: procesos de instauración gradual, que afectan a múltiples troncos nerviosos y que se caracterizan por ser simétricos y generalizados, con afectación preferentemente distal.")
6
CONCEPTOS Radiculopatías: trastornos que afectan a las raíces nerviosas Poliradiculopatía: afectación de múltiples raíces nerviosas de forma consecutiva Miopatía:Cambio o falla funcional (anormalidad) del músculo. La mayoría se presenta con debilidad simétrica y proximal de las extremidades con reflejos y sensación preservados.
 del músculo. La mayoría se presenta con debilidad simétrica y proximal de las extremidades con reflejos y sensación preservados.")
10
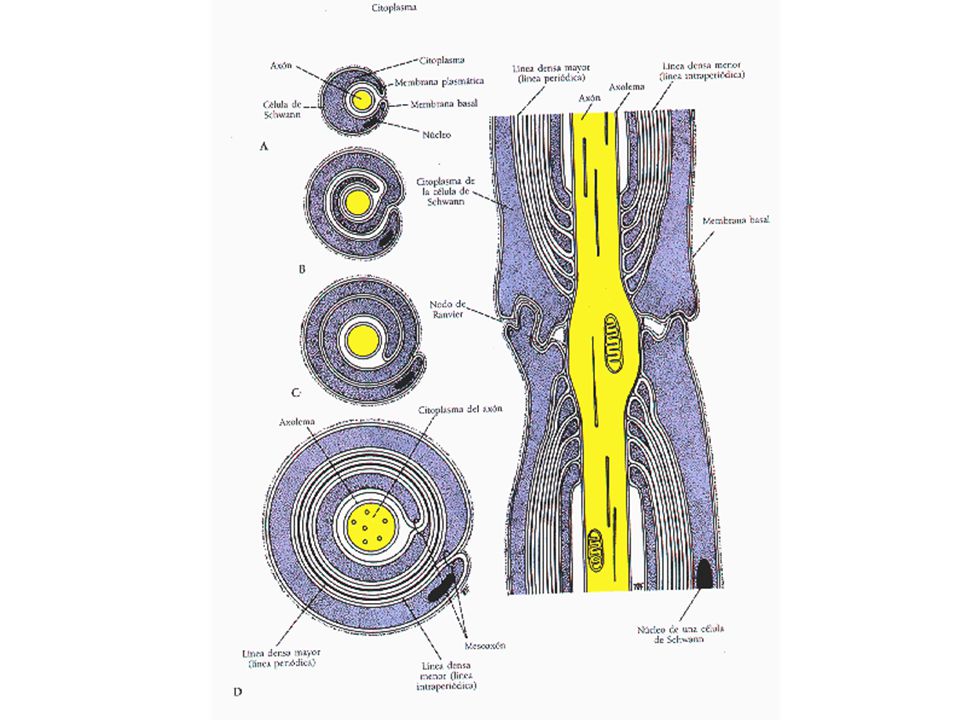
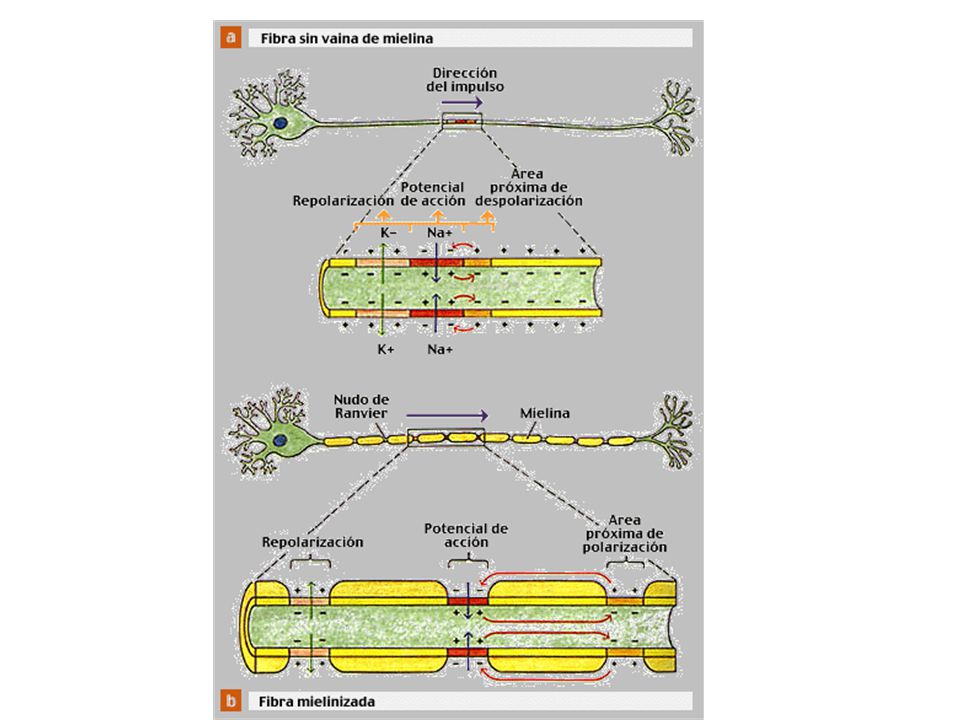
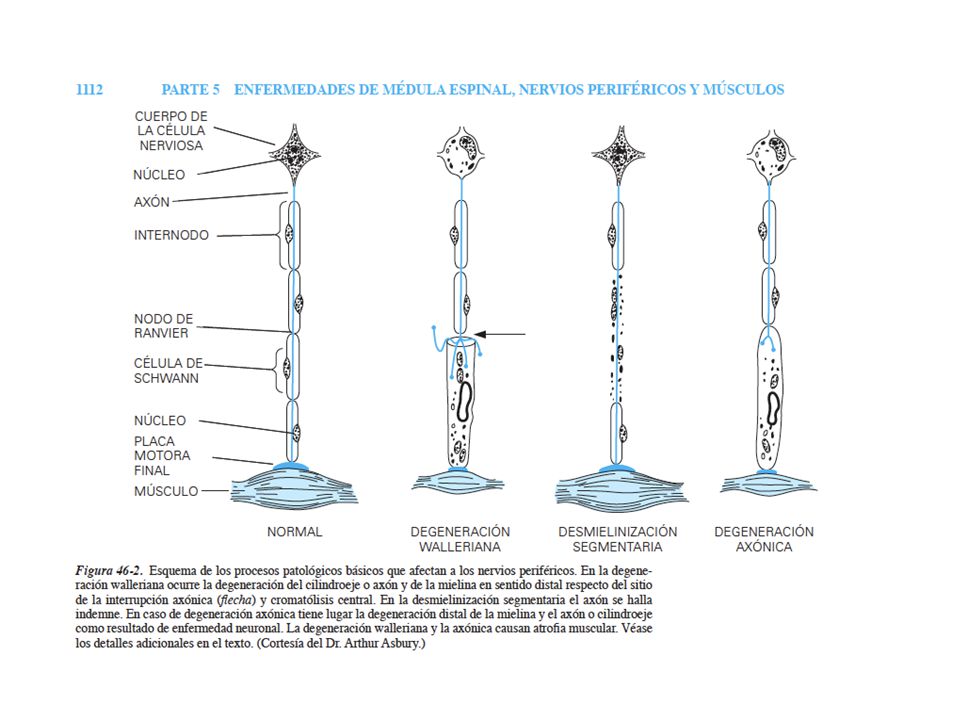
PROCESOS PATOLÓGICOS BÁSICOS QUE AFECTAN A LOS NERVIOS PERIFÉRICOS
Degeneración walleriana Se rompe la estructura entre el axón y su vaina de mielina Desmielinización segmentaria Degeneración focal de la vaina de mielina con axón indemne Quedan expuestos los segmentos del axón Degeneración axónica

12
Clasificación de la neuropatías según su ubicación
polineuropatía plexopatía Mononeuropatía múltiple No es neuropatía -simétrica -distal en media-guante -dolorosa o no sensitivomotora -hiporreflexia asimétrica -Asimétrica -principio doloroso -múltiples nervios en una sola extremidad -inicio rápido de debilidad y atrofia -Arreflexia aislada -Distribución por nervio -antecedentes: DM, vasculitis, presión -dolorosa o no Hiporreflexia aislada -signos de neurona motora superior (hiperreflexia) -daño vesical e intestinal marcado -síntomas unilaterales (brazo, pierna, cara) -hiperventilación
 -daño vesical e intestinal marcado. -síntomas unilaterales (brazo, pierna, cara) -hiperventilación.")
13
Clasificación de neuropatías según el tipo de fibra
Sensitiva, fibras pequeñas (neuropatías dolorosas y pérdida sensitiva disociada) -Neuropatías sensitivas hereditrias (tempranas) -lepra lepromatosa -neuropatía diabética de fibras pequeñas -amiloidosis -apolipoproteinemia Enfermedad de Fabry -Neuropatía por VIH Sensitiva de fibras grandes -Síndrome de Sjörgen -neuropatía por hipovitaminosis B12 -Neuropatía por cisplatino -Envenenamiento por piridoxina -Ataxia de Friederich Fibras pequeñas y grandes (pérdida sensitiva global) -Neuropatía sensitiva carcinomatosa -Neuropatías sensitivas hereditarias -neuropatía sensitiva diabética -Intoxicación por vacor -neuropatía xantomatosa de la cirrosis biliar primara (tabes dorsal) Neuropatías de predominio motor -Neuropatías inmunitarias agudas (Guillain-Barré) -neuropatías motoras-sensitivas hereditarias -Porfiria intermitente aguda -nueropatía diftérica -Neuropatía por plomo -Neuritis braquial -Neuropatía diabética del plexo lumbosacro Neuropatías autonómicas -Aguda -crónica
 -Neuropatías sensitivas hereditrias (tempranas) -lepra lepromatosa. -neuropatía diabética de fibras pequeñas. -amiloidosis. -apolipoproteinemia. Enfermedad de Fabry. -Neuropatía por VIH. Sensitiva de fibras grandes. -Síndrome de Sjörgen. -neuropatía por hipovitaminosis B12. -Neuropatía por cisplatino. -Envenenamiento por piridoxina. -Ataxia de Friederich. Fibras pequeñas y grandes (pérdida sensitiva global) -Neuropatía sensitiva carcinomatosa. -Neuropatías sensitivas hereditarias. -neuropatía sensitiva diabética. -Intoxicación por vacor. -neuropatía xantomatosa de la cirrosis biliar primara (tabes dorsal) Neuropatías de predominio motor. -Neuropatías inmunitarias agudas (Guillain-Barré) -neuropatías motoras-sensitivas hereditarias. -Porfiria intermitente aguda. -nueropatía diftérica. -Neuropatía por plomo. -Neuritis braquial. -Neuropatía diabética del plexo lumbosacro. Neuropatías autonómicas. -Aguda. -crónica.")
15
SÍNTOMAS DE LA ENFERMEDAD NERVIOSA PERIFÉRICA
Perturbación de la función motora Deterioro persistente de la función motora (días – meses) El grado de debilidad es proporcional al número de axones o motoneruonas afectadas Polineuropatías: distribución simétrica de la debilidad o la parálisis , empieza sobre todo en pies y piernas
 El grado de debilidad es proporcional al número de axones o motoneruonas afectadas. Polineuropatías: distribución simétrica de la debilidad o la parálisis , empieza sobre todo en pies y piernas.")
16
Reflejos tenidinososos disminuidos Hipoestesia
Se afecta la sensibilidad en forma simétrica en los segmentos distales de las extremidades (más en piernas que en brazos) Parestesias, dolor y disestesias Particularmente intensos en manos y pies, “alfilerazos”, punzadas, hormigueo toques Ataxia sensitiva y temblor Deformidad y cambios tróficos Pies, manos y columna vertebral, sobre todo cuando la enfermedad comienza en la infancia Trastornos vegetativos Anhidridosis e hipotensión ortostática
 Parestesias, dolor y disestesias. Particularmente intensos en manos y pies, alfilerazos , punzadas, hormigueo toques. Ataxia sensitiva y temblor. Deformidad y cambios tróficos. Pies, manos y columna vertebral, sobre todo cuando la enfermedad comienza en la infancia. Trastornos vegetativos. Anhidridosis e hipotensión ortostática.")
17
SÍNDROME DE GUILLAIN-BARRÉ
“POLINEUROPATÍA INFLAMATORIA AGUDA” Polirradiculoneuropatía desmielinizante aguda de origen inmunológico Afecta preferentemente a adultos jóvenes Más de 2/3 tienen antecedentes de infección viral respiratoria o gastrointestinal Grupo herpes (CMV) También se asocia a procedimientos quirúrgicos, linfomas y LES
 También se asocia a procedimientos quirúrgicos, linfomas y LES.")
18
SÍNDROME DE GUILLAIN BARRÉ
Autoinmune La desmielinización se produce por un doble mecanismo, mediada por linfocitos y por anticuerpos circulantes Se caracteriza por la presencia de inflamación, desmielinización y degeneración axonal, restringida al SNP Desmielinización segmentaria y multifocal Puede haber degeneración axonal secundaria al proceso de desmielinización en las zonas mas intensas de inflamación

19
SÍNDROME DE GUILLAIN BARRÉ
CLÍNICA Tetraparesia flácida y arreflexia Escasos síntomas sensitivo Normamente no hay afectación esfinteriana La debilidad suele iniciar en los miembros inferiores y asciende progresivamente Puede haber parestesias distales (pero no hay pérdida marcada de la sensibilidad) Síntomas autonómicos: taquicardia, hipotensión postural, hipertensión La debilidad se establece en un lapso por lo general de horas a pocos días pero puede ser hasta en 28 días y no remite hasta que alcanza la debilidad máxima, a la que sigue una meseta de tiempo variable
 Síntomas autonómicos: taquicardia, hipotensión postural, hipertensión. La debilidad se establece en un lapso por lo general de horas a pocos días pero puede ser hasta en 28 días y no remite hasta que alcanza la debilidad máxima, a la que sigue una meseta de tiempo variable.")
20
CRITERIOS DIAGNÓSTIVOS PARA EL SGB
NECESARIOS 1. Debilidad prgresiva en 2 o más miembros oir neuropatía 2. Arreflexia 3. Evolución de la enfermedad <4 semanas 4. Exclusión de otras causas DE APOYO 1. Debilidad relativamente simétrica 2. Afección sensitiva de grado leve 3. Afección del nervio facial o de toros pares craneales 4. Ausencia de fiebre 5. Perfil tipico en el LCR (acelular, aumento en el valor de proteínas) 6. Signos electrofisiológicos de desmielinización
 6. Signos electrofisiológicos de desmielinización.")
21
TRATAMIENTO DE SGB Medidas generales Medidas farmacológicas
Buen estado hidroelectrolítico y nutricional, controlar los procesos infecciosos agregados, prevenir trombosis venosa profunda, psicoterapia de apoyo Medidas farmacológicas Plasmaféresis (para formas graves) inmunoglobulina
 inmunoglobulina.")
22
POLINEUROPATÍA DESMIELINIZANTE INFLAMATORIA CRÓNICA
Pico de incidencia: 5ª y 6ª década, varones afectados más frecuentemente 1/3 antecedentes de indección Predilección por los nervios proximales y las raíces espinales Inicio similar al síndrome de Guillain-Barré pero con instauración de los síntomas más gradual (a veces >2 meses)
")
23
PDIC Curso crónico progresivo o con caídas intermitentes
Mayor afectación sensitiva que en el SGB. Debilidad en los músculos proximales y distales mientras que las parestesias son de predominio distal Afectación de pares craneales bajos y de músculos intercostales TRATAMIENTO Corticoides, plasmaféresis, inmunoglobulinas Casos refreactarios inmunosupresores

24
NEUROPATÍA DIABÉTICA 2 tipos: -simétricas: daño de las fibras pequeñas -asimétricas: inicio agudo: radiculoneuropatía diabética troncal, neuropatía diabética de raíces y plexo lumbosacro, neuropatía oculomotora (causas vasculares) inicio gradual: neuropatía del mediano de la muñeca, cubital del codo, peronea en la cabeza del peroné y del cutáneo lateral del muslo (por compresión) *Lo normal es que se presente una combinación de ambas
 inicio gradual: neuropatía del mediano de la muñeca, cubital del codo, peronea en la cabeza del peroné y del cutáneo lateral del muslo (por compresión) *Lo normal es que se presente una combinación de ambas")
25
NEUROPATÍA DIABÉTICA SIMÉTRICA
Afecta a más del 50% de los px diabéticos que tienen más de 25 años de evolución Combinación de degeneración axonal (preferentemente distal) y desmielinización segmentaria El dolor y la insensibilidad en las extremidades predisponen a formación de úlceras en los pies 2 posibles causas: El aumento de la concentración neuronal de glucosa disminuye el descenso de diacilglicerol, cinasa de proteína C y actividad de la Na,K triptofosfatasa pérdida de axones y demielinización Disminución del flujo sanguíneo en los vasa nervorum Existen 4 tipos
 y desmielinización segmentaria. El dolor y la insensibilidad en las extremidades predisponen a formación de úlceras en los pies. 2 posibles causas: El aumento de la concentración neuronal de glucosa disminuye el descenso de diacilglicerol, cinasa de proteína C y actividad de la Na,K triptofosfatasa pérdida de axones y demielinización. Disminución del flujo sanguíneo en los vasa nervorum. Existen 4 tipos.")
26
a) Polineuropatía sensitiva distal
Es la forma más frecuente de polineuropatía diabética Parestesias e hipoestesias en guante y calcetín, pérdida de la sensibilidad vibratoria y arreflexia distal (fibras gruesas) Dolor con sensación quemante en los pies que empeora notablemente por las noches (fibras pequeñas)
 Dolor con sensación quemante en los pies que empeora notablemente por las noches (fibras pequeñas)")
27
b) Neuropatía autonómica
Se asocia a neuropatía sensitiva y cursa con clínica cardiovascular (hipotensión ortostática, taquicardia en reposo), genitourinaria (vejiga neurógena, impotencia) y gastrointestinal (gastroparesia, vómitos) Diarrea es el síntoma intestinal más frecuente
, genitourinaria (vejiga neurógena, impotencia) y gastrointestinal (gastroparesia, vómitos) Diarrea es el síntoma intestinal más frecuente.")
28
c) Neuropatía dolorosa aguda
Dolor quemante muy intenso en las plantas de los pies acompañando de hipersensibilidad cutánea No se afectan miembros superiores No hay déficit motor

29
d) Neuropatía motora proximal de miembros inferiores
“síndrome de Garland” Amiotrofía diabética Dolor lumbar bajo y áreas glúteas, seguido de debilidad progresiva de cuadríceps e iliopsoas con eventual atrofia y pérdida de los reflejos

30
POLINEUROPATÍAS ASIMÉTRICAS
Menos frecuentes que las simétricas, pueden aparecer en el curso de las polineuropatías simétricas Patogenia con frecuencia vascular NEUROPATÍAS CRANEALES Daño de los pares craneales Nervios oculomotores, con mayor frecuencia VI, III y rara vez el IV Por lo general px >50 años Parálisis del VI (abductor) diplopia indolora repentina, recuperación espontánea Parálisis del III par repentina, a veces precedida por dolor retroorbitario intenso que puede durar varios días, hay diplopia, ptosis unilateral y limitacion de la mirada medial y superior Parálisis del VII (facial) parálisis de Bell
 diplopia indolora repentina, recuperación espontánea. Parálisis del III par repentina, a veces precedida por dolor retroorbitario intenso que puede durar varios días, hay diplopia, ptosis unilateral y limitacion de la mirada medial y superior. Parálisis del VII (facial) parálisis de Bell.")
31
NEUROPATÍAS DE LAS EXTREMIDADES
Por atrapamiento Predisposición a compresión por edema del endoneuro y factores vasculares Tratamiento quirúrgico NEUROPATÍAS DEL TRONCO Afectación aguda y dolorosa unilateral de uno o más nervios torácicos Más frecuente en >50 años Dolor y disestesias unilaterales en tórax y abdomen Puede confundirse con herpes

32
UNIÓN NEUROMUSCULAR Unidad motora: conjunto de fibras musculares inervadas por una misma neurona motora

33
UNIÓN NEUROMUSCULAR

34
MIASTENIA GRAVIS Enfermedad de la unión neuromuscular
Trastorno neuromuscular caracterizado por debilidad y fatiga de los músculos esqueléticos Disminución del número de receptores de acetilcolina en las uniones neuromusculares debido a un proceso autoinmunitario Afecta más frecuentemente a mujeres entre la 2º y 3º década (hombres entre 4ª y 5ª/ 6ª y 7ª décadas) 1/10000 personas DEBILIDAD Y FATIGABILIDAD MUSCULAR
 1/10000 personas. DEBILIDAD Y FATIGABILIDAD MUSCULAR.")
35
PATOGENIA DE MG Anticuerpos vs receptores nicotínicos de acetilcolina que: Bloquean el receptor de acetilcolina Degradación acelerada de la acetilcolina Activan el depósito de complemento sobre la membrana postináptica, con la consiguiente destrucción de los receptores y el aplanamiento a largo plazo de los pliegues del receptor postsináptico no hay contracción muscular

36
3 características marcan el diagnóstico de miasteina gravis:
Carácter fluctuante de la debilidadm empeora despues del ejercicio y mejora con el reposo o sueño Afectación de la musculatura craneal, preferentemente extraocular, con ptosis y diplopía Respuesta clínica a los fármacos colinérgicos (anticolinesterásicos)
")
37
MANIFESTACIONES CÍNICAS DE MG
Debilidad y fatigabilidad muscular La debilidad aumenta con la actividad repetida y puede mejorar con el reposo o el sueño Distribución: músculos craneales (labios y extraoculares suelen ser los primeros) y a menoduo diplopía y ptosis suelen ser los síntomas iniciales Expresión de gruñido cuando el px intenta sonreir Debilidad en la masticación Cambios en la voz Puede haber dificultad para la deglución o del paladr, lengua o faringe No hay alteraciones sensitivas, autonómicas ni pupilares Debilidad de miembros proximal y asimétrica
 y a menoduo diplopía y ptosis suelen ser los síntomas iniciales. Expresión de gruñido cuando el px intenta sonreir. Debilidad en la masticación. Cambios en la voz. Puede haber dificultad para la deglución o del paladr, lengua o faringe. No hay alteraciones sensitivas, autonómicas ni pupilares. Debilidad de miembros proximal y asimétrica.")
38
CRISIS MIASTÉNICA: cuando la debilidad muscular respiratoria produce
MIASTENIA GRAVE OCULAR: solo síntomas oculares por 2-3 años (es probable que no se generalice) CRISIS MIASTÉNICA: cuando la debilidad muscular respiratoria produce
 CRISIS MIASTÉNICA: cuando la debilidad muscular respiratoria produce.")
39
Prueba de edrofonio (tensilon)
60% a 95% en pacientes con OMG y 72% a 95% en GMG 60% to 95% of patients with OMG and in 72% to 95% with GMG also seen in congenital myasthenic syndromes, the Lambert-Eaton syndrome, intracranial aneurysms, brainstem lesions, cavernous sinus tumors, end-stage renal disease, and in muscle diseases neostigmine

40
Tratamiento MG

41
Miopatías

42
MIOPATÍAS ENFERMEDADES DEL MÚSCULO Inflamatorias Distrofias musculares
Miopatías distales Miopatías miotónicas Miopatías congénitas Miopatías mitocondriales Miopatías metabólicas

43
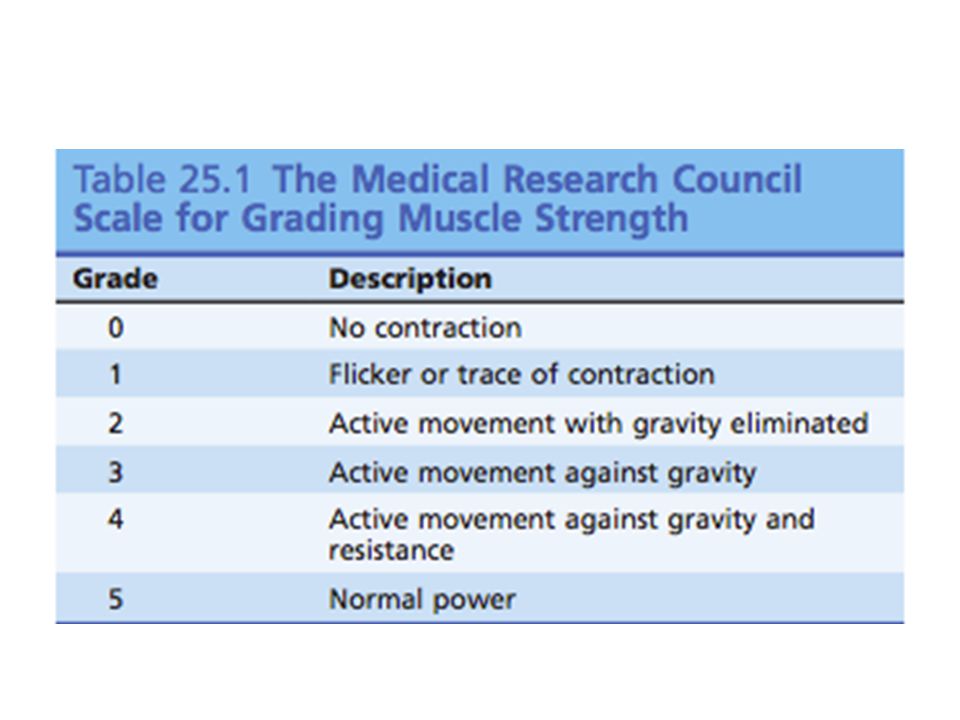
Datos clínicos Debilidad muscular:
Debilidad intermitente: MG, parálisis periódica, deficiencia metabólica de la glicolisis, de la utilización de ácidos grasos y miopatías mitocondriales. Debilidad persistente: distrofia muscular, polimiositis y dermatomiositis. Debilidad facial y alas escapulares (distrofia fascioescapulohumeral) Debilidad facial distal de miembros y mitononia de agarre (distrofia miotonica tipo I) Debilidad de músculos d elso nervios craneales (UNM, DMOF, MM, MC) Atrofia y debilidad del antebrazo flexor y del quadriceps asimétrico, (miositis de cuerpos de inclusión o miopatías distales.) Analizar sindrome de la cabeza caída. (MG, ALS, Nemalina M, HPT, FM, IBM) Debilidad de las extremidades dsitales (MD)
 Debilidad facial distal de miembros y mitononia de agarre (distrofia miotonica tipo I) Debilidad de músculos d elso nervios craneales (UNM, DMOF, MM, MC) Atrofia y debilidad del antebrazo flexor y del quadriceps asimétrico, (miositis de cuerpos de inclusión o miopatías distales.) Analizar sindrome de la cabeza caída. (MG, ALS, Nemalina M, HPT, FM, IBM) Debilidad de las extremidades dsitales (MD)")
44
Datos de debilidad muscular
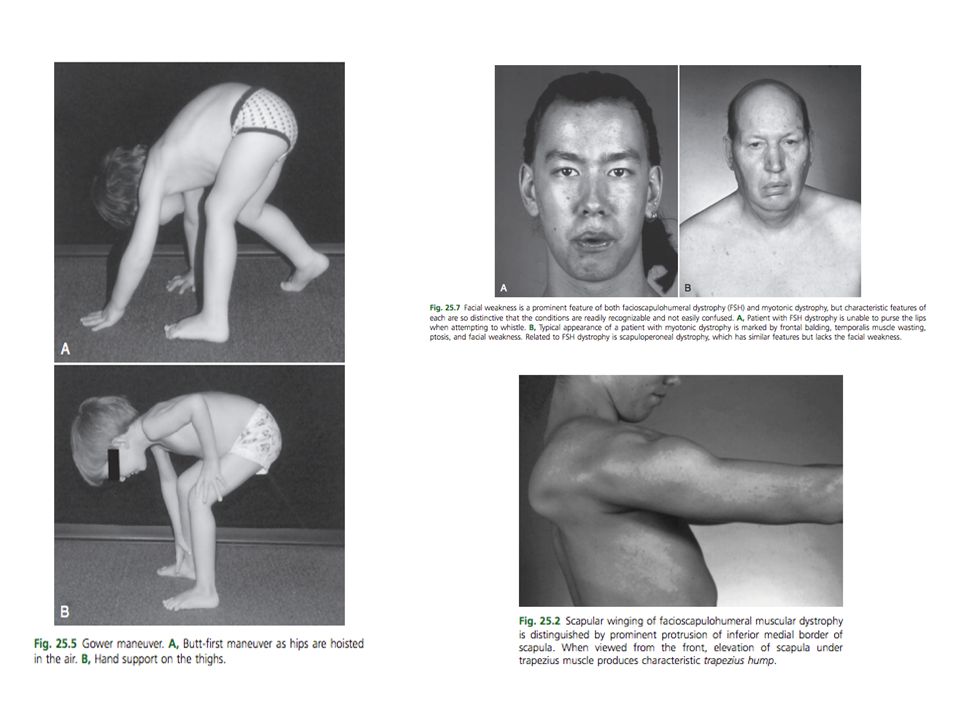
Falla funcional (Incapacidad) Debilidad muscular Cerrar los ojos a la fuerza Fascial superior Levantar la cabeza en posición prona Extensores del cuello Levantar la cabeza en psoción supina Flexores del cuello Levantar los brazos por encima de la cabeza Proximales del brazo Genu recurvatum Extensores de la rodilla Caminar de puntillas Tendon de aquiles Marcha equina Compartimiento anterior de la pierna Marcha de pato Músculos de la cadera Arruga de la cara Facial inferior Levantarse del piso sin subir las extremidades (signo de Gowers) Músculos cadera, muslo y tronco Levantarse de una silla sin utilizar los brazos músculos de la cadera
 Debilidad muscular. Cerrar los ojos a la fuerza. Fascial superior. Levantar la cabeza en posición prona. Extensores del cuello. Levantar la cabeza en psoción supina. Flexores del cuello. Levantar los brazos por encima de la cabeza. Proximales del brazo. Genu recurvatum. Extensores de la rodilla. Caminar de puntillas. Tendon de aquiles. Marcha equina. Compartimiento anterior de la pierna. Marcha de pato. Músculos de la cadera. Arruga de la cara. Facial inferior. Levantarse del piso sin subir las extremidades (signo de Gowers) Músculos cadera, muslo y tronco. Levantarse de una silla sin utilizar los brazos músculos de la cadera.")
48
Fatiga: inabilidad para mantener o soxtener una fuerza (transmisión neuromuscular, alteración producción de energía y miopatía crónica) Astenia : falta de energía o cansancio extremo.

49
Dolor muscular, calambres y rigidez.
Fibromialgia, polimialgia reumática y dolor traumático. Calambre o espasmo muscular: enfermedad de la neurona motora, radiculopatías, polineuropatías o DMD. Contractura muscular: desordenes glicoliticos de falla de energía, no se relaja, silencio EMG, Emery Dreifuss MD y miopatía de Bethlem. Contracción dolorosa, involuntaria y localizada con endurecimientoñ. De inicio abrupto de corta duración.

50
Rigidez muscular: Síndrome de la persona rígida, neuromiotonia y miocimia
Miotonia: De acción o percusión. DM1 (distal), DM2 (proximal), miotonia congénita (Cl), paramiotonia congénita, canalopatías de sodio. Atrofia e hipertorfia: Disfernolopatías y FSHD. Granulomas sarcoides, infecciones, deposito amiloide, miositis focal. Descarga neuronas motoras espinales. Contraccione sinvolunatarias principalmente tronco y extremidad inferior. Marcha dura y rigida hiperlordosis movmientos y soidos subitos. Hiperexcetibilidad nervios perifericos. Fasciulaciones y ondulaciones d elso músculos. No relajación rigidez y sudoración. Canale spotasio activados por voltaje. Contrcción prolongada seguido de reljación lenta. Rigidez.
, DM2 (proximal), miotonia congénita (Cl), paramiotonia congénita, canalopatías de sodio. Atrofia e hipertorfia: Disfernolopatías y FSHD. Granulomas sarcoides, infecciones, deposito amiloide, miositis focal. Descarga neuronas motoras espinales. Contraccione sinvolunatarias principalmente tronco y extremidad inferior. Marcha dura y rigida hiperlordosis movmientos y soidos subitos. Hiperexcetibilidad nervios perifericos. Fasciulaciones y ondulaciones d elso músculos. No relajación rigidez y sudoración. Canale spotasio activados por voltaje. Contrcción prolongada seguido de reljación lenta. Rigidez.")
51
Laboratorio Enzimas séricas: CK (CK-MM) Sin provocación, truama menor, espasmo prolongado o crisis. AST, ALT, aldolasa y GGT(hígado). Análisis DNA Duchenne y Becker. Deleción, duplicación

52
Examen de ejercicios del antebrazo.
Catéter vena antecubutal Muestra de sangre de base para amonia y ácido lactico. Abrir y cerrar la mano por un minuto. Obtener sangre en los minutos 1,2,4,6 y 10. Deficiencia de miofosforilasa o miodenilato deaminasa

53
Biopsia de músculo Cuadriceps o biceps
Células inflamatorias endomisiales (poliomiositis) Con fibras con vacuolas bordeadas, depositos amiloides e inclusiones TDP-43 (miositis de cuerpos de inclusión) Atrofia perifascicular con inflamación perivascular y perimisial (dermatomiositis) Microscopia electrónica, inmunohistoquimica y western blot.
 Con fibras con vacuolas bordeadas, depositos amiloides e inclusiones TDP-43 (miositis de cuerpos de inclusión) Atrofia perifascicular con inflamación perivascular y perimisial (dermatomiositis) Microscopia electrónica, inmunohistoquimica y western blot.")
62
Tratamiento miopatías
Prednisona retrasa DM de Duchenne por 3 años. DM1 y DM2, fármacos antiomiotonicos (fenitoína y mexiletina) junto con marcapasos. Paralisis periódica hipocalemica KCL en manitol cada 30 minutos. Tratar enfermedad tiroidea o infecciosa originante. Retirar fármacos causantes en las inducidas por los mismos. En el resto vigilar y tratar condiciones asociadas.
 junto con marcapasos. Paralisis periódica hipocalemica KCL en manitol cada 30 minutos. Tratar enfermedad tiroidea o infecciosa originante. Retirar fármacos causantes en las inducidas por los mismos. En el resto vigilar y tratar condiciones asociadas.")
63
Miopatías inflamatorias
Dermatomiusocitis signo de Gotrron

64
Polimiositis e IBM Se presenta en adultos. C8/C4
Poco dolor, malestar, fiebre y anorexia. Diágnostico de exclusión Sobrediagnosticada IBM mayores de 50 mas comun, distal asimetrica

65
Tratamiento PM y DM: Glucocorticoides
Otros fármacos inmunosupresivos (azatripiona, metrotexate o micofenolato) Inmunomodulación con inmunoglobulina IV IBM resitente a terapias inmunosupresivas.
 Inmunomodulación con inmunoglobulina IV. IBM resitente a terapias inmunosupresivas.")
66
ALS (Esclerosis Lateral Amiotrófica)
Denervación y atrofia muscular con gliosis fibrilar columnas laterales. Daño por glutamato (riluzole) (SOD, TDP43) Clinica depende de singos de neurona motora alta o baja. Tendra los dos, progresiva debilitante. Es de exclusión. Incidencia 1-3 por 100 mil, prevalencia 3-5 por 100 mil Sobreviven 3-5 años Alta Dificultad masticar, tragar mov. cara y lengua. Hiperactividad refleja y resistencia espástica Disrtria y afecto pseudobulbar Baja Debilidad asimetrica de comienzo distal. Calambres en la mañana y atrofia muscular. Fasciculaciones tempranas y predominancia extensora d ela mano. Denervación y atrofia. Gliosis firbirlar columnas laterales. Glutamato daño, SOD, TDP43
 (SOD, TDP43) Clinica depende de singos de neurona motora alta o baja. Tendra los dos, progresiva debilitante. Es de exclusión. Incidencia 1-3 por 100 mil, prevalencia 3-5 por 100 mil. Sobreviven 3-5 años. Alta. Dificultad masticar, tragar mov. cara y lengua. Hiperactividad refleja y resistencia espástica. Disrtria y afecto pseudobulbar. Baja. Debilidad asimetrica de comienzo distal. Calambres en la mañana y atrofia muscular. Fasciculaciones tempranas y predominancia extensora d ela mano. Denervación y atrofia. Gliosis firbirlar columnas laterales. Glutamato daño, SOD, TDP43.")
67
Síndrome de Eaton Lambert
Forma especial de miastenia Relacionado con carcinoma de células pequeñas Afecta con mayor frecuencia a los hombres (5:1)
")
68
Respuesta autoinmune contra canales de calcio presinápticos
Anticuerpos circulantes que interfieren con la descarga de Ach en los sitios muscarínicos y nicotínicos Defecto en la descarga de Ach desde las terminaciones nerviosas presinápticas Respuesta autoinmune contra canales de calcio presinápticos Incremento de la fuerza con una serie de contracciones voluntarias en ausencia de miotonía Se caracteriza por bloqueo presináptico de la liberación de Ach y rpoduce el defecto opuesto de transmisión neuromuscular al que se registra en la miastenia grave Pérdida de canales de calcio sensbiles al voltaje sobre la terminación nervioso motora presináptica. Los canales de Ca están enlazados de manera cruzada y agregados por los autoanticuerpos del tipo IgG, con reducción final en el número de canales funcionales Aparecen anticuerpos contra un componente específico de la membrana presináptica que tienen el efecto de disminuir la liberación presináptica de acetilcolina lo contrario a la miastenia!

69
Diagnóstico Disautonomía que se caracteriza por sequedad de boca, impotencia, dificultad para orinar y estreñimiento Diferencial Miastenia grave Miopatía de cuerpos de inclusión Poliomiositis

70
Primeros síntomas Dificultad para levantarse de la silla, subir escaleras, caminar Puede haber un incremento temporal de la fuerza muscular Reflejos tendinosos disminuidos Debilidad proximal de extremidades pélvicas Otras: brazos, diplopía, ptosis, disartría

71
Estudios electrodiagnósticos
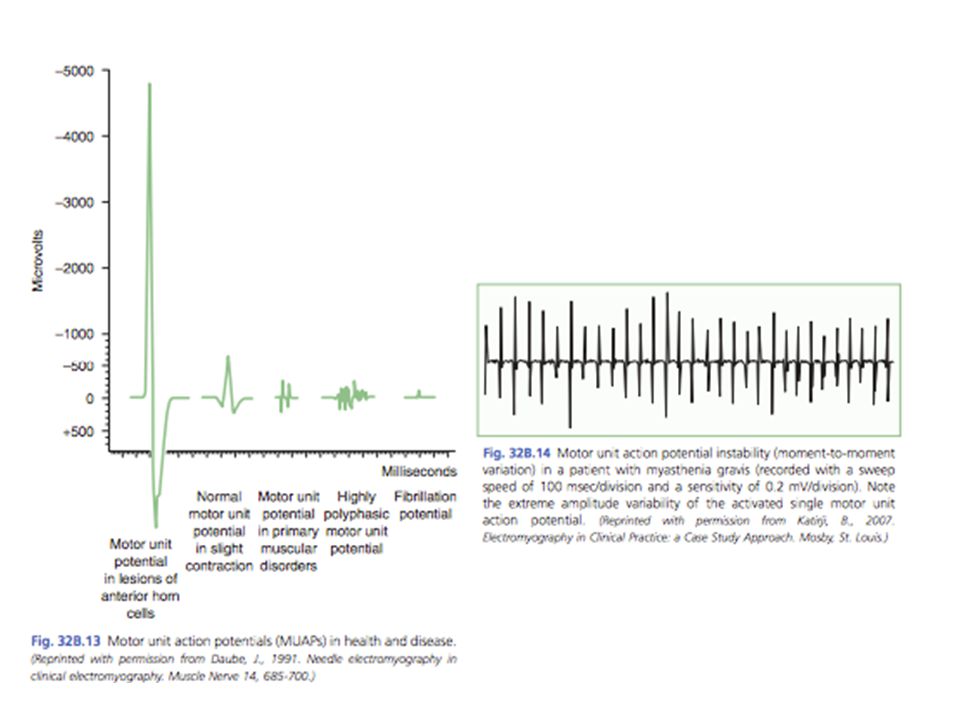
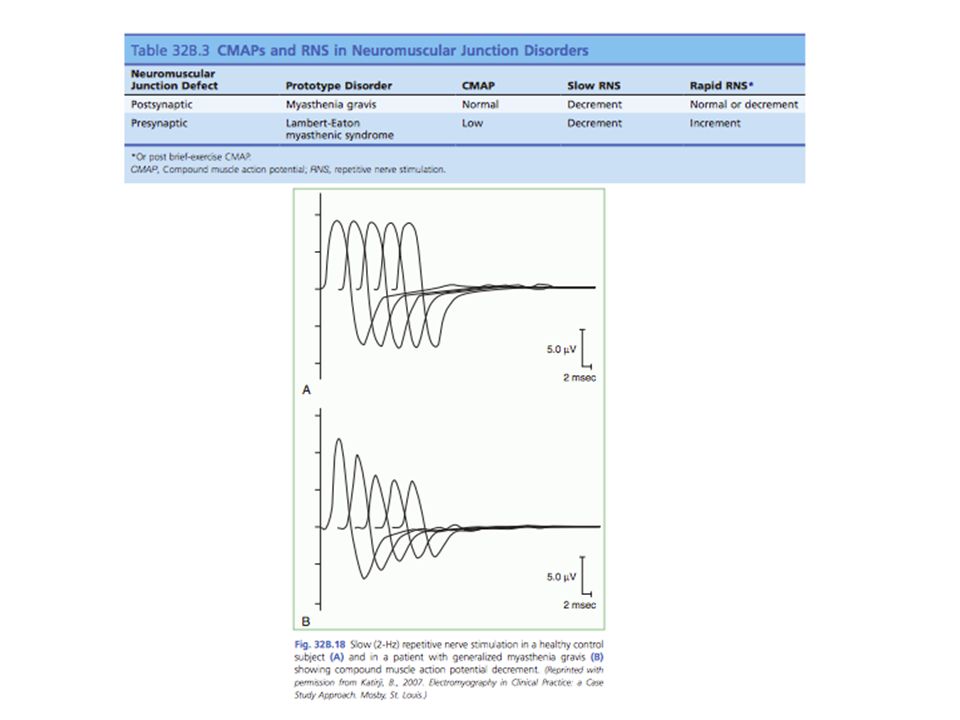
Potencial de acción muscular de baja amplitud En miastenia grave, el potencial es normal o se acerca Reacción de incremento Rápida estimulación + contracción voluntaria = amplitud de los potenciales de acción

72
Otros estudios Método serológico para anticuerpos Buscar un tumor
Tomografía con emisión de positrones

73
Tratamiento 3,4-diaminopiridina (3,4-DAP) Bloque canales de potasio
Prolonga despolarización e intensifica la liberación de vesículas acetilcolínicas Prednisona + azatioprina en días alternos En NO tumorales Plasmaféresis + prednisona y azatioprina

74
Anticuerpos contra el nervio donde Ach es liberada
Eaton-Lambert Miastenia Gravis Anticuerpos contra el nervio donde Ach es liberada Anticuerpos contra el músculo receptor de ACh Empieza en las extremidades y se desarrollo hacia arriba Empieza en los ojos y se desarrolla hacia abajo La debilidad mejora con la actividad La debilidad empeora con la actividad Se asocia a carcinoma de células pequeñas Se asocia a timoma Tratamiento: aminopiridinas Tratamiento: inhibidores de acetilcolinesterasa Agegar werkicke hauffman, paralisis de bell, dushenne, becker

75
Neurona motora alta Esclerosis lateral primaria: debilidad espastica progresiva con disfagia y disartria. (degeneracion corticoespinal y –bulbar) No hay datos de denervacion en EMG. Sobrevida 3 años. Paraplegia familiar espástica: autosomica dominante, tercera o cuarta decada, debilidad espástica progresiva distal de extremidad baja (espastina). Recesiva corticoespinal y dorsal.
. Recesiva corticoespinal y dorsal.")
76
Neurona motora baja Enfermedad de Kennedy: debilidad progresiva y atrofia de extremidades y bulbar. Insensibilidad androgena. Masculinos adultos. Ausencia de espasticidad y neuropatia sensorial leve. CAG en cromosoma X.

77
Distrofia muscular de duchenne

78
La más frecuente y mejor conocida de las distrofias musculares
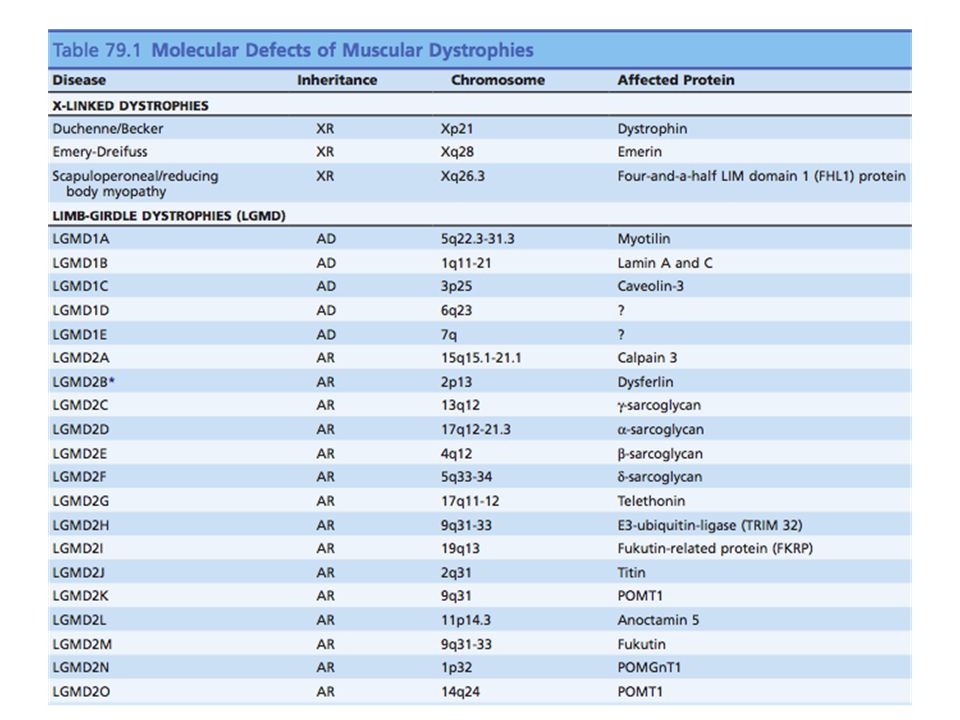
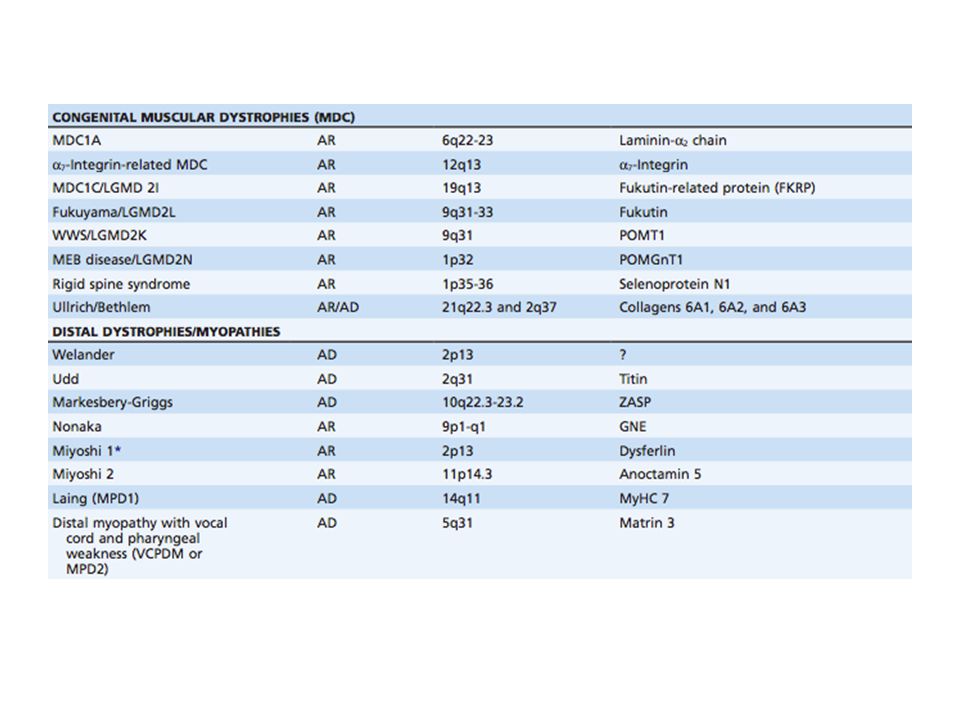
Inicia en la infancia y sigue una evolución progresiva y rápida 13-33 / 100,000 en un año 1 / 3300 varones vivos Recesivo ligado al X

79
Aspectos clínicos Suele reconocerse entre el tercer año de vida
Antes de empezar a caminar Retraso psicomotor Elevación de CK Lordosis y marcha de ganso Inicio: iliopsoas, cuadríceps y glúteos Músculos pretibiales Cintura escapular y extremidades superiores después que pelvicocrurales Algunos pacientes cuando logran caminar o correr, parecen ser menos activos que lo ordinario y son susceptibles a las caídass. Su dificultad creciente para caminar, correr y subir escaleras, la lordosis y la marcha de ganso son aún más manifiestas conforme el tiempo pasa. Músculos pretibiales: caída del pie y marcha sobre las puntas Los músculos serratos, las partes bajas de los pectorales, dorsales anchos, bíceps y supinadores largos quedan dañados más o menos en ese orden

80
Pueden incrementar pero acaban decreciendo
Excepto gemelos, vastos laterales y deltoides Músculos aumentados “de caucho” Menos fuertes y más hipotónicos Duchenne “Hércules Farnese” Aumento de las pantorrillas, sólo los gemelos y menos grado los vastos laterales y deltoides, son sostenidamente grandes De caucho cosistencia firme y resistente

81
Aumento de las pantorrillas, sólo los gemelos y menos grado los vastos laterales y deltoides, son sostenidamente grandes De caucho cosistencia firme y resistente Los músculos de la cintura pélvica, la columna lumbosacra y los hombros se debilitan y agotan. La debilidad de los músculos abdominales y paravertebrales explica la postura lordótica y el abdomen protuberante cuando el sujeto adopta la bipedestación, y el dorso es redondeado cuando está sentado La debilidad bilateral de los extensores de las rodillas y las caderas interfiere con el equilibrio y las actividades, como subir escaleras o levantarse de una silla o desde una postura inclinada Cuando se encuentra en bipedestación y caminar, el paciente coloca los pies bastante separados con objeto de incrementar su base de apoyo. Para incorporarse desde la posición sedante, flexiona primero el tronco hacia arriba al deslizar las manos por los muslos también hacia arriba. Cuando se levanta del suelo posición de cuatro puntos mediante la extesión de los barzos y las piernas hasta la ascensión mayor posible y en seguida desplaza de manera alternativa la mano sobre el muslo correspondiente Gowers Balanceo debilidad de ambos glúteos Muchos niños caminan sobre sus dedos del pie por la contractura en los gemelos Los ánguglos escapulares, se observan por arriba de los hombros cuando se mira de frente por el debilitamiento delos músculos que fijan las escápulas contra el tórax serrato mayor, romboides y la parte baja del trapecio

82
Extremidades flácidas
Inclinación pélvica, lordosis Lordosis lumbar, flexión y abducción de caderas, flexión de rodillas y flexión plantar Postura: Reflejos tendinosos atenuados Huesos delgados y desmineralizados Problemas cardíacos Infecciones pulmonares Insuficiencia respiratoria Muerte Por lo regular las extremidades están flácidas, pero conforme la incapacidad progresa, aparecen contracturas fibrosa Terminan con inclinación pélvica y lordosis compensatoria par aconservar el equilibrio durante la bipedestación. Postura ordinaria: lordosis lumbar, flexión y abudcción de las caderas, flexión de roddilas y flexión plantar Los reflejos tendinosos están atenuados y luego se pierden. El ultimo que se pierde es el del tobillo Huesos delgados y desmineralizados. Problemas cardíacos (pérdida de fibras y restitución por fibrosas) Muerte por infecciones pulmonares e insuf resp, alguna veces por descompensación cardíaca. No más del 20-25% está vivo después de los 25 años
 Muerte por infecciones pulmonares e insuf resp, alguna veces por descompensación cardíaca. No más del 20-25% está vivo después de los 25 años.")
83
Distrofia muscular de becker

84
3-6 por 100,000 varones nacidos vivos
Ligado a X que se limita a varones Debilidad e hipertrofia como en Duchenne Inicio más tardío Media: 12 años Límites: 5-45 años Afección al corazón menos frecuente Se relaciona con la de Duchenne

85
Patología de las distrofias
Duchenne Degeneración segmentaria y fagocitosis provenientes de las fibras musculares Necrosis excita un proceso regenerativo o de restauración Hipertrofia Por aumento de tamaño de las fibras sanas Seudohipertrofia Sustitución lipocítica de fibras musculares degeneradas Degeneración segmentaria y fagocitosis provenientes de las fibras musculares únicas o grupos de fibras La necrosis excita un proceso regenerativo o de restauración deformidad de las fibras en bifurcacion. La hipertrofia es por el aumento de tamaño de las fibras sanas inducido por el trabajo ante la lesión de las fibras adyacentes La seudohipertrofia se debe a sustitución lipocítica de fibras musculares degeneradas La hipertrofia real cambia a una forma de seudohipertrofia

86
Causas Sarcolema se puede romper más fácil o desgarrAse
Gen anormal en el cromosoma X y su producto génico distrofina Ausente en los pacientes con el fenotipo de Duchenne Estructura anormal en el tipo de Becker Pérdida de distrofina origina una desaparicIón de PRD y la disrupción del complejo distroglucano-proteína Sarcolema se puede romper más fácil o desgarrAse Gen anormal en el cromosoma X y su producto génico distrofina La distrofina está ausente en los pacientes con el fenotipo de Duchenne, se encuentra pero con estructura anormal en el tipo de Becker Duchenne y Becker y sus formas intermedias se conocen como distrofinopatías Imagen En los músculos cardíacos y esquelético la distrofina se encuentra en la superficie citoplásmica del sarcolema, donde interactúa con la actina F del citoesqueleto. También se encuentra enlazada con un complejo de proteínas del sarcolema que se conocen como proteínas y glucoproteínas relacionadas con la distrofina PRD y GRD la pérdida de distrofina origina una desaparicón de PRD y la disrupción del complejo distroglucano-proteína. sarcolema se puede romper más fácil o desgarrse

87
Diagnóstico Análisis del gen de distrofina en DNA
Leucocitos o de 50 mg de músculo de fibra estriada Inmunotinción ELISA

Presentaciones similares