Descargar la presentación
La descarga está en progreso. Por favor, espere

1
CROMATOGRAFÍA Y COMPONENTES DE UN SISTEMA CROMATOGRÁFICO
Catedrática: Dra. Silvia Echeverría, Ph.D., Depto. Fisicoquímica Facultad de Ciencias Químicas y Farmacia, USAC. GUATEMALA, C.A.

2
FUNDAMENTOS DE SEPARACIÓN
PRINCIPIOS GENERALES TÉCNICAS DE SEPARACIÓN TEORÍA MECANISMOS DE SEPARACIÓN

3
Mezclas y la necesidad de métodos de separación
Complejidad de las muestras: Sangre, agua de río, alimentos licuados, entre otros. Las muestras pueden contener mezclas de solutos, micelas, coloides y otras partículas en suspensión.

4
Técnicas de separación clásicas
Separación de componentes de interés Precipitación Destilación Extracción Soxhlet

5
Extracción con ultrasonido (USE)
Extracción con disolventes Extracción con ultrasonido (USE) (Líquido-líquido) Extracción en fase sólida (SPE)
 (Líquido-líquido) Extracción en fase sólida (SPE)")
6
Extracción con Líquidos presurizados (PLE)
Extracción asistida con microondas (MAE)
")
7
CROMATOGRAFÍA Nombre genérico asignado a muchas y distintas técnicas de separación. El nombre se debe al botánico ruso Mikhail Tsweet, quién acuñó el término a principios de la década de 1900, cuando logró separar varios pigmentos de plantas tales como xantofilas y clorofilas, al hacer pasar soluciones de esta mezcla por columnas de vidrio empacadas con carbonato de calcio finamente dividido. Cada pigmento recorrió la columna con diferente velocidad y terminó apareciendo cada componente como bandas de color. Chroma, palabra griega para color y Graphein que significa escribir.

8
CLASIFICACIÓN DE LOS MÉTODOS CROMATOGRÁFICOS
Basada en la forma en que se ponen en contacto la FASE MÓVIL y la FASE ESTACIONARIA: En COLUMNA y en PLANO. Basada en el tipo de FASE MÓVIL: CG, CL, CFS.

9
Todas las formas de cromatografía se basan en la separación de muestras de mezclas al entrar en contacto con dos fases: fase móvil y fase estacionaria. Una de las fases se mueve en relación con la otra. Los componentes de la muestra se distribuyen entre las dos fases de acuerdo con sus solubilidades (o afinidades) relativas hacia ellas. Los componentes que no interaccionan con la fase estacionaria pasan rápidamente con la fase móvil. Al revés, los componentes que si interaccionan con la fase estacionaria, se mueven con mayor lentitud.

10
Los componentes de la muestra se mueven con velocidades determinadas por los tiempos relativos que pasan en las fases móvil y estacionaria. A su vez, dichos tiempos quedan determinados por los coeficientes de partición para cada componente entre las dos fases. De esta forma, se pueden separar distintos componentes debido a la velocidad relativa con que atraviesan la fase estacionaria.

11
Se puede realizar la cromatografía de elusión (con dos fases líquidas) en varias formas distintas. En muchos casos la fase estacionaria es un disolvente (por ejemplo agua) adsorbido y por consiguiente, inmovilizado en un soporte sólido, como papel de celulosa, o sílice en una columna empacada. La muestra que se va a resolver o separar en sus partes componentes se disuelve en un pequeño volumen de un segundo disolvente que tendrá el papel de fase móvil. A continuación se agrega la solución de la muestra al soporte sólido (que contiene la fase estacionaria inmovilizada).
 adsorbido y por consiguiente, inmovilizado en un soporte sólido, como papel de celulosa, o sílice en una columna empacada. La muestra que se va a resolver o separar en sus partes componentes se disuelve en un pequeño volumen de un segundo disolvente que tendrá el papel de fase móvil. A continuación se agrega la solución de la muestra al soporte sólido (que contiene la fase estacionaria inmovilizada)..")
12
A medida que la fase móvil atraviesa la fase estacionaria, los solutos se distribuyen entre las fases de acuerdo con sus coeficientes de partición respectivos. Se pueden agregar más fases móviles para ELUIR los componentes de la fase estacionaria a distintas velocidades. (Puede imaginarse este tipo de cromatografía como una serie continua de extracciones líquido-líquido). Al final todos los componentes serán eluidos del sistema del que se trate y así la mezcla quedará separada.
. Al final todos los componentes serán eluidos del sistema del que se trate y así la mezcla quedará separada.")
13
TEORÍA DE LAS SEPARACIONES CROMATOGRÁFICAS Coeficientes de partición
Todas las separaciones cromatográficas están gobernadas por coeficientes de partición KD de solutos, entre las fases estacionaria y móvil. Para un soluto S se establece un equilibrio dinámico, esto es: S móvil S estacionario

14
KD = [S]est/[S]móv (croma 01)
El coeficiente de partición o de distribución, KD, es igual a la relación de la concentración del soluto en las dos fases, tal como en la ecuación: KD = [S]est/[S]móv (croma 01) Donde KD es el coeficiente de partición y [S]est y [S]móv son las concentraciones molares del soluto S en las fases estacionaria y móvil respectivamente. En el caso ideal, el valor de KD debe permanecer constante dentro de un amplio margen de concentraciones de soluto, para asegurar que la relación misma de [S] est y [S] móv quede constante. Se puede suponer que la cromatografía efectuada bajo estas condiciones tiene comportamiento lineal, lo cuál permite hacer determinaciones cuantitativas a partir del cromatograma.
![KD = [S]est/[S]móv (croma 01)](http://slideplayer.es/slide/5909943/18/images/14/KD+%3D+%5BS%5Dest%2F%5BS%5Dm%C3%B3v+%28croma+01%29.jpg "El coeficiente de partición o de distribución, KD, es igual a la relación de la concentración del soluto en las dos fases, tal como en la ecuación: KD = [S]est/[S]móv (croma 01) Donde KD es el coeficiente de partición y [S]est y [S]móv son las concentraciones molares del soluto S en las fases estacionaria y móvil respectivamente. En el caso ideal, el valor de KD debe permanecer constante dentro de un amplio margen de concentraciones de soluto, para asegurar que la relación misma de [S] est y [S] móv quede constante. Se puede suponer que la cromatografía efectuada bajo estas condiciones tiene comportamiento lineal, lo cuál permite hacer determinaciones cuantitativas a partir del cromatograma.")
15
En la práctica, bajo la mayor parte de condiciones normales de trabajo, se puede suponer que KD es constante, aunque a concentraciones muy altas la fase estacionaria puede volverse parcial o totalmente saturada.

16
TIEMPO DE RETENCIÓN TIEMPO DE RETENCIÓN
En esta figura se observa un cromatograma característico para una muestra que contiene un solo analito. El tiempo que transcurre luego de la inyección de la muestra hasta que se alcanza la concentración máxima del analito en el detector, se denomina Tiempo de Retención (tR)
")
17
El tiempo que tarda un analito en eluirse de una columna, también se suele llamar tiempo de retención o tiempo de residencia. Si un soluto en la fase móvil no interacciona en absoluto con la fase estacionaria, se moverá con la misma velocidad que la fase móvil, dicho tiempo se puede representar como tmóv y también se le denomina, en algunas ocasiones, tiempo muerto (tM). Si el analito pasa una parte del tiempo en la fM y otra parte en la fE, su velocidad de avance estará determinada por el coeficiente de partición KD y en consecuencia, distintos analitos serán eluidos de la columna en tiempos distintos que dependen de sus coeficientes de partición dentro de la columna.
. Si el analito pasa una parte del tiempo en la fM y otra parte en la fE, su velocidad de avance estará determinada por el coeficiente de partición KD y en consecuencia, distintos analitos serán eluidos de la columna en tiempos distintos que dependen de sus coeficientes de partición dentro de la columna.")
18
La velocidad lineal promedio del movimiento, u, de la fase móvil, se puede expresar de acuerdo a la ecuación: u = L/tmóv (croma 02) En donde L es la longitud de la columna. En forma similar, para cualquier pico cromatográfico, se puede expresar la velocidad lineal promedio de migración del soluto por medio de la ecuación: ṽ = L/tR (croma 03) El tiempo de retención tR de un soluto se puede relacionar con su coeficiente de partición KD expresando la velocidad de migración del soluto ṽ en función de una fracción de la velocidad de la fase móvil, tal como en la ecuación:
 En donde L es la longitud de la columna. En forma similar, para cualquier pico cromatográfico, se puede expresar la velocidad lineal promedio de migración del soluto por medio de la ecuación: ṽ = L/tR (croma 03) El tiempo de retención tR de un soluto se puede relacionar con su coeficiente de partición KD expresando la velocidad de migración del soluto ṽ en función de una fracción de la velocidad de la fase móvil, tal como en la ecuación:")
19
ṽ = u * (fracción del tiempo en la fase móvil)
(croma 04) Sin embargo, se deben tomar en cuenta los volúmenes de las fases estacionaria y móvil, a los cuáles se les llama Vest y Vmóv respectivamente. Basados en lo anterior, la velocidad de migración del soluto para determinado pico cromatográfico, se puede expresar así: ṽ = u * (1)/(1+KD Vest/Vmóv) (croma 05)
 Sin embargo, se deben tomar en cuenta los volúmenes de las fases estacionaria y móvil, a los cuáles se les llama Vest y Vmóv respectivamente. Basados en lo anterior, la velocidad de migración del soluto para determinado pico cromatográfico, se puede expresar así: ṽ = u * (1)/(1+KD Vest/Vmóv) (croma 05)")
20
k’ = (tR – tmóv)/tmóv (croma 06)
EL FACTOR DE CAPACIDAD Es un parámetro para comparar las velocidades relativas de migración de soluto en las columnas. Dicho factor k’ se puede calcular con la ecuación: k’ = (tR – tmóv)/tmóv (croma 06) En el caso ideal, k’ debe estar dentro del intervalo de 1 a 5. Si k’ es mucho menor que 1, la elución será tan rápida que será difícil determinar con exactitud los tiempos de retención. Por lo contrario, si el factor capacidad es mucho mayor que 20, los tiempos de retención serán excesivos.
/tmóv (croma 06) En el caso ideal, k’ debe estar dentro del intervalo de 1 a 5. Si k’ es mucho menor que 1, la elución será tan rápida que será difícil determinar con exactitud los tiempos de retención. Por lo contrario, si el factor capacidad es mucho mayor que 20, los tiempos de retención serán excesivos.")
21
Ejemplo para calcular k’:
Si el tiempo de retención tR de un pico cromatográfico es 65 y tmóv es 30s, calcular k’, el factor de capacidad. Método: Calcular k’ de acuerdo con la ecuación: k’= (tR – tmóv)/tmóv k’ = 65 – 30/30 = 1.17 s-¹ EL FACTOR DE SELECTIVIDAD , para dos solutos A y B, se define como = K’B/K’A (croma 07) donde KB es el coeficiente de distribución para la especie más fuertemente retenida y KA es el factor de distribución para la menos retenida.
/tmóv. k’ = 65 – 30/30 = 1.17 s-¹. EL FACTOR DE SELECTIVIDAD , para dos solutos A y B, se define como. = K’B/K’A (croma 07) donde KB es el coeficiente de distribución para la especie más fuertemente retenida y KA es el factor de distribución para la menos retenida.")
22
De acuerdo con esta definición, siempre será mayor que la unidad.
Sustituyendo una de las ecuaciones anteriores y la análoga para la especie B en la última ecuación, se obtiene, después de reordenar, una relación entre el factor de selectividad para dos analitos y sus factores de retención: = k’B/´k’A (croma 08) Donde k’B y k’A son los factores de retención de A y de B respectivamente. La sustitución en la ecuación croma 06, para las dos especies en la ecuación croma 07 da otra ecuación que permite la determinación de a partir de un cromatograma experimental:
 Donde k’B y k’A son los factores de retención de A y de B respectivamente. La sustitución en la ecuación croma 06, para las dos especies en la ecuación croma 07 da otra ecuación que permite la determinación de a partir de un cromatograma experimental:")
23
= [(tR)B – tmóv]/[(tR)A – tmóv] (croma 09)
EFICIENCIA de las COLUMNAS CROMATOGRÁFICAS Se puede describir la eficiencia de una columna ya sea en función de El número de platos teóricos N, o de la Altura de plato H (HETP). Ambas acepciones se relacionan con la ecuación de Van Deemeter, en honor del primer científico que describió teóricamente una ecuación para cuantificar la eficiencia de las columnas cromatográficas.
![ = [(tR)B – tmóv]/[(tR)A – tmóv] (croma 09)](http://slideplayer.es/slide/5909943/18/images/23/%EF%81%A1+%3D+%5B%28tR%29B+%E2%80%93+tm%C3%B3v%5D%2F%5B%28tR%29A+%E2%80%93+tm%C3%B3v%5D+%28croma+09%29.jpg "EFICIENCIA de las COLUMNAS CROMATOGRÁFICAS. Se puede describir la eficiencia de una columna ya sea en función de. El número de platos teóricos N, o. de la Altura de plato H (HETP). Ambas acepciones se relacionan con la ecuación de Van Deemeter, en honor del primer científico que describió teóricamente una ecuación para cuantificar la eficiencia de las columnas cromatográficas.")
24
Ecuación de Van Deemeter: H = L/N
H es HETP L es longitud de la columna

25
La terminología de platos teóricos y altura de plato para describir la eficiencia de las columnas usadas en cromatografía es un legado histórico de un modelo teórico de cromatografía que ya se ha superado en gran parte. El uso del término “Plato teórico” no debe concebirse como una representación física de la columna ni de su operación, más bien es un parámetro arbitrario para describir su eficiencia. La eficiencia de la columna mejora a medida que: a) Aumenta el número de platos y b) Se reduce la altura de plato. El número de platos teóricos puede variar desde algunos cientos hasta varios cientos de miles, mientras que la altura de plato puede variar desde algunos milímetros hasta algunas decenas de micras.
 Aumenta el número de platos y b) Se reduce la altura de plato. El número de platos teóricos puede variar desde algunos cientos hasta varios cientos de miles, mientras que la altura de plato puede variar desde algunos milímetros hasta algunas decenas de micras.")
26
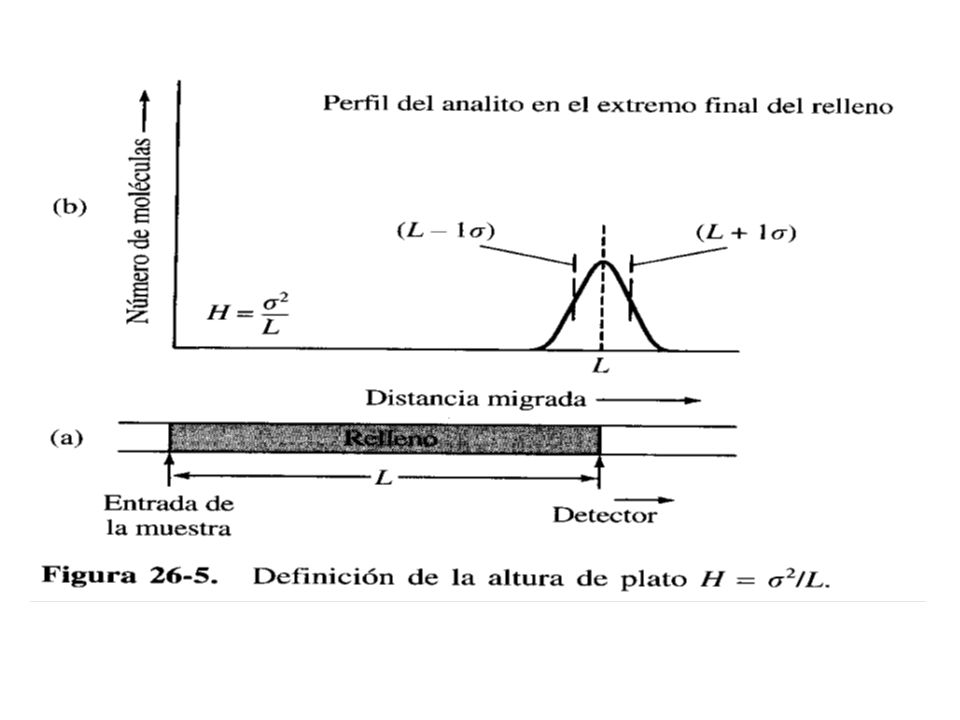
Se ha observado que un pico cromatográfico se ensancha a medida que aumenta su tiempo de retención
Asimismo el tiempo de retención de un pico aumentará si se incrementa la longitud de la columna. Entonces, a medida que la longitud de la columna aumenta, los picos cromatográficos se ensanchan. Ya que el pico tiene la forma de una curva de distribución de Gauss, se puede expresar esa forma en función del ancho que abarca más o menos una desviación estándar σ. Entonces se puede expresar la desviación estándar en función de la varianza, σ² H = σ² /L (croma 10) Notar que las unidades de L son cm, las de σ² son cm², debido a lo cuál H también tiene cm como unidades y se puede concebir como relacionada con la longitud de
 Notar que las unidades de L son cm, las de σ² son cm², debido a lo cuál H también tiene cm como unidades y se puede concebir como relacionada con la longitud de.")
28
la columna que contiene a (L – σ) proporción del analito.
También se puede obtener el número de platos teóricos en forma directa, a partir de un cromatograma, ya que se puede demostrar que: N= 16(tR/w)² (croma 11) Donde tR es el tiempo de retención y w es el ancho de la base del pico cromatográfico. Tal como se ha observado, el tiempo de retención de un soluto, se relaciona con la longitud de la columna. En la práctica es más fácil medir tiempos de retención en forma directa en cromatogramas y usarlos para expresar la eficiencia de la columna.
² (croma 11) Donde tR es el tiempo de retención y w es el ancho de la base del pico cromatográfico. Tal como se ha observado, el tiempo de retención de un soluto, se relaciona con la longitud de la columna. En la práctica es más fácil medir tiempos de retención en forma directa en cromatogramas y usarlos para expresar la eficiencia de la columna.")
29
EJEMPLO PARA CALCULAR LA ALTURA DE PLATO TEÓRICO HETP:
Un pico cromatográfico tiene un tiempo de retención de 52 s. El ancho de la base del pico equivale a 3.2 s, en la intersección de los lados de pico con la línea base. Si la columna tiene 500 cm de longitud, calcular la HETP en centímetros por plato. Procedimiento: Calcular N con la ecuación N =16(tR/w)² y luego calcular HETP con la ecuación HETP = L/N N = 16 (52/3.2)² = 16 (16.25)² = 4225 Por lo que: HETP = 500/4225 = 0.12 cm por plato
² y luego calcular HETP con la ecuación HETP = L/N. N = 16 (52/3.2)² = 16 (16.25)² = Por lo que: HETP = 500/4225 = 0.12 cm por plato.")
30
FORMAS DE LOS PICOS CROMATOGRÁFICOS
En esta figura se observan los perfiles de concentración de los analitos A y B en dos tiempos distintos en su migración a lo largo de la columna.

31
Se puede apreciar que en el caso típico, los picos tienen la forma de una curva de Gauss de distribución normal. Las formas de dichos picos se pueden atribuir al movimiento aleatorio de las partículas de soluto, a medida que la solución atraviesa la columna. Habrá un tiempo de retención que corresponda a la cantidad máxima de moléculas de soluto que eluyen de la columna. Las moléculas de soluto sufren muchos miles de transferencias entre las fases móvil y estacionaria por las que atraviesan dentro de la columna; sin embargo el tiempo que tarda una molécula en cada fase es aleatorio e impredecible. El soluto sólo se puede mover mientras está en la fase móvil y así se concluye que si la molécula de soluto pasa más tiempo en la fase estacionaria, recorre la columna con más lentitud y por lo tanto,

32
Si la molécula pasa mayor proporción de tiempo en la fase móvil, recorrerá la columna más rápidamente. El intercambio de un soluto entre una y otra fase requiere gasto de energía y como en cualquier sistema en el que sucede una transferencia de energía, el proceso es de naturaleza aleatoria. El resultado de estos intercambios aleatorios entre las dos fases produce el ensanchamiento de los picos cromatográficos. El ancho de un pico se relaciona con el tiempo promedio que tarda un soluto en eluirse de la columna, o sea, con su tiempo de retención. Los picos cromatográficos con mayor tiempo de retención también son los más anchos.

33
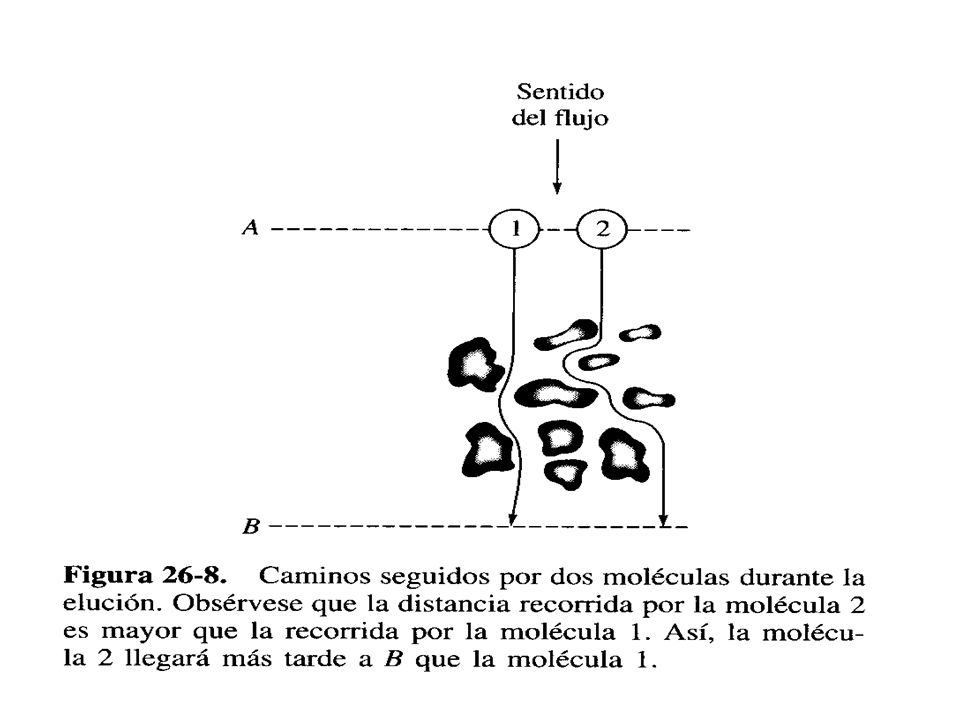
ENSANCHAMIENTO DE BANDA O DE PICO
Comúnmente al fenómeno de ensanchamiento de los picos cromatográficos se le llama ensanchamiento de banda. Dicho ensanchamiento se debe a varias causas, Efectos de la distribución de la fM dentro de la columna: El flujo es más rápido en el centro debido a efectos de resistencia (o de fricción) en las paredes, y dicho efecto se acentúa mientras más larga sea la columna. Difusión de las moléculas dentro de la fM al pasar a lo largo de la fE: Dado que la concentración de una especie será mayor en el centro que en las paredes de la columna debido a los efectos de distribución del flujo, entonces las moléculas tienden también a
 en las paredes, y dicho efecto se acentúa mientras más larga sea la columna. Difusión de las moléculas dentro de la fM al pasar a lo largo de la fE: Dado que la concentración de una especie será mayor en el centro que en las paredes de la columna debido a los efectos de distribución del flujo, entonces las moléculas tienden también a.")
34
Difundirse hacia las orillas, debido a un gradiente de concentración
Difundirse hacia las orillas, debido a un gradiente de concentración. Este proceso continuará mientras haya separación cromatográfica, a lo largo de la columna. c. Difusión secundaria o parásita de los solutos: ya que muchas columnas contienen partículas de materiales en la fE, algunas moléculas de soluto se moverán en línea más recta que otras por la columna, pues pueden tener más desviaciones en su camino. Esto a su vez causará que algunas moléculas eluyan con más rapidez que otras y de esa forma se ensanchará la banda cromatográfica. d. La presencia de pequeñas zonas dentro de la columna que contengan fM “estancada”, debido al uso de empaque con materiales porosos donde la fM no pueda pasar con facilidad, puede causar también ensanchamiento de bandas.

36
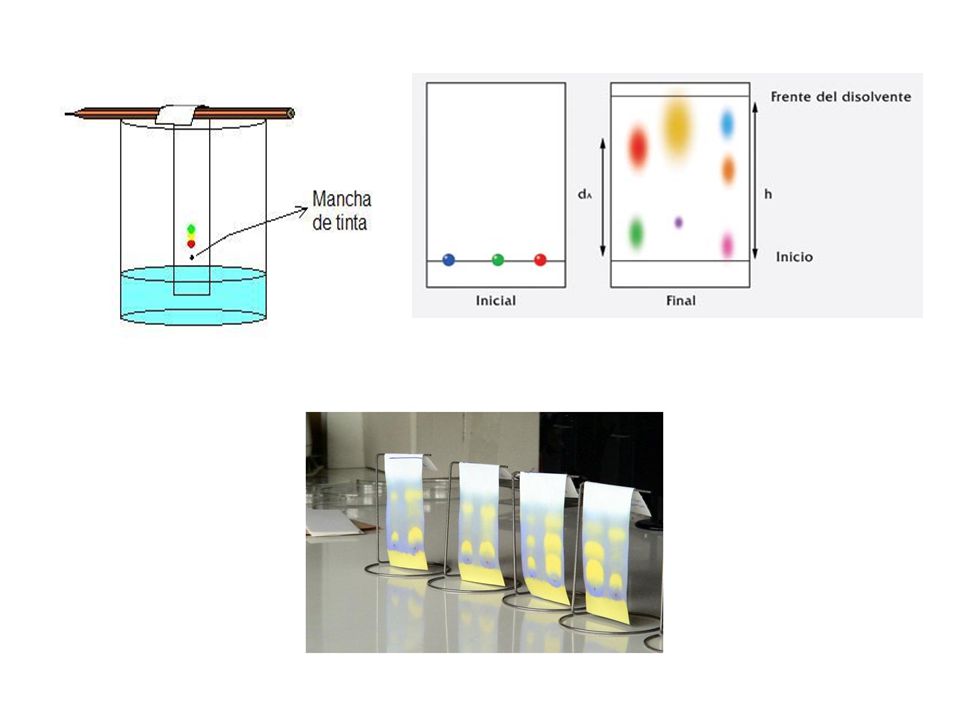
CROMATOGRAFÍA EN PAPEL
Las fibras de celulosa del papel pueden actuar como fase estacionaria directamente, o ser un soporte para la adsorción de una fase estacionaria líquida, como el agua, por ejemplo. En el caso común, un extremo del papel se sumerge una pequeña distancia en la cámara cromatográfica que contiene a la fM. La mezcla en solución se depositó previamente en forma de una mancha en la línea de inicio. La mancha se aplicó lo más concentrada y pequeña posible, para evitar la dispersión prematura de la muestra por lo que normalmente se siembran manchas muy pequeñas, una sobre la otra, una vez que la anterior se haya secado por completo. La fM subirá por el papel por capilaridad. Se debe colocar la tapa de la cámara para asegurar que la atmósfera que rodee al papel esté saturada con el vapor de la fM.

38
Justo antes de que el frente del solvente llegue a la orilla superior del papel, se traza otra línea con lápiz, para marcar la distancia recorrida por la fM. A continuación se saca el papel y se deja secar. Luego ya se pueden calcular los valores de Rf (Factor de retención) para cada componente, como la relación de la distancia recorrida por la mancha entre la distancia recorrida por el frente del solvente desde la marca de inicio. Rf = dist recorrida por el analito/dist recorrida por el frente del solvente Debido a que los valores de Rf no son reproducibles con más de 2 cifras significativas, pues dicho valor presenta sensibilidad a variables tales como el grosor de la fE, la humedad que contiene la fM entre otros,

39
se recurre a otro valor que resulta más representativo, conocido como Factor de Retención Relativo Rx, el cuál se define así: Rx = Dist recorrida por el analito/dist recorrida por una sustancia de referencia La sustancia de referencia es generalmente un patrón interno, el cuál se define como una sustancia de identidad conocida y con alta pureza que se emplea para fines de comparación. El Rx ideal es igual a 1, pues así se confirmaría la identidad de un analito con respecto de la sustancia de referencia.

40
CROMATOGRAFÍA EN CAPA FINA: (Thin Layer Chromatography, TLC)
Conceptualmente es muy similar a la cromatografía en papel, pero permite hacer mejores separaciones y tiende a mostrar un comportamiento más reproducible. Para la TLC, se usa como fase estacionaria un sólido finamente dividido (inmovilizado sobre un vidrio, o sobre una hoja de material polimérico o de aluminio), que puede ser alúmina (Al2O3), gel de sílice, entre otros. La fase móvil puede ser agua, solución acuosa de amoníaco o alguna otra mezcla, por ejemplo, una solución de alcohol/agua/ácido acético.
, que puede ser alúmina (Al2O3), gel de sílice, entre otros. La fase móvil puede ser agua, solución acuosa de amoníaco o alguna otra mezcla, por ejemplo, una solución de alcohol/agua/ácido acético.")
41
Las placas de TLC se marcan y se revelan en forma muy similar a la que se usa para la cromatografía en papel, pero teniendo el cuidado de trazar las líneas con el lápiz (con mina de grafito) en forma delicada para evitar que la fase estacionaria de la placa se fracture, y esto afecte la separación. Con frecuencia se recubre la fase estacionaria con un material fluorescente para ayudar a visualizar los componentes a medida que se separan, siempre que los compuestos en estudio posean electrones que al ser excitados emitan fluorescencia que se evidenciará al observarlos con luz ultra violeta. También se pueden usar reactivos reveladores nebulizados sobre las placas para hacer visibles a los analitos ya separados.

42
La forma de aplicar las muestras también es similar a la utilizada en cromatografía en papel, aplicando manchas pequeñas y secándolas antes de volver a aplicarlas para concentrarlas.

45
CROMATOGRAFÍA EN COLUMNA
(Estudiar la presentación del grupo de Edy Jiménez)
")
46
CROMATOGRAFÍA DE GASES
En la cromatografía de gases, los componentes de una muestra vaporizada se separan como consecuencia de su distribución o reparto entre una fase móvil gaseosa y una fase estacionaria sólida o líquida mantenida dentro de una columna. Para efectuar la separación en fase gaseosa, la muestra líquida se introduce en el inyector, en donde se convierte en vapor, antes de pasar a la columna cromatográfica. La elución se consigue mediante el flujo constante de una fase móvil de gas inerte, conocido como gas portador,

47
O por su nombre en el idioma inglés: Carrier Gas
O por su nombre en el idioma inglés: Carrier Gas. A diferencia de otros tipos de cromatografía, la fM no interacciona con las moléculas del analito, por lo cuál su única función consiste en transportar o acarrear al analito a lo largo de la columna Se utilizan dos tipos de cromatografía gaseosa: a) Cromatografía de Gas-sólido (CGS) b) Cromatografía de Gas-líquido (CGL) En CGS la fM es un gas y la fE es un sólido que retiene a los analitos por adsorción física. Este tipo de cromatografía permite separar y determinar gases de bajo peso molecular, como los componentes del aire, sulfuro de hidrógeno (H2S), monóxido de carbono (CO) y óxidos de nitrógeno.
 Cromatografía de Gas-sólido (CGS) b) Cromatografía de Gas-líquido (CGL) En CGS la fM es un gas y la fE es un sólido que retiene a los analitos por adsorción física. Este tipo de cromatografía permite separar y determinar gases de bajo peso molecular, como los componentes del aire, sulfuro de hidrógeno (H2S), monóxido de carbono (CO) y óxidos de nitrógeno.")
48
Sin embargo, esta variante de cromatografía ha tenido aplicación limitada debido a la retención semipermanente de moléculas activas o polares y a que a los picos de elución suelen presentar grandes “colas” como consecuencia de la naturaleza no lineal del proceso de elución. De manera que esta técnica no ha tenido una aplicación generalizada, salvo la separación de ciertas especies gaseosas de bajo peso molecular, que ya han sido mencionadas. La Cromatografía de GAS-LÍQUIDO, se basa en la distribución o reparto del analito entre una fM gaseosa y una fase líquida inmovilizada sobre la superficie de un sólido inerte o en las paredes de un tubo capilar. En 1941 Martin y Synge propusieron por primera vez el concepto de la CGL. En 1955 apareció en el mercado el primer equipo comercial para CGL.

49
A partir de entonces, el crecimiento de la aplicación de esta técnica ha sido espectacular, pues desde su introducción comercial han ocurrido muchos cambios y mejoras en los instrumentos. En la década de 1970 se hicieron comunes los integradores electrónicos y los equipos de procesamiento de datos por computadora. En las décadas de 1980 y 1990 se introdujo el uso de computadoras para el control automático de los parámetros instrumentales, tales como la temperatura de la columna, las velocidades de flujo y la inyección de la muestra, también se desarrollaron instrumentos de alto rendimiento a costo moderado y lo más importante: el desarrollo de columnas tubulares abiertas que permiten la separación de los componentes de mezclas complejas en períodos de tiempo breves.

50
COMPONENTES BÁSICOS DE UN CROMATÓGRAFO DE GASES
Gas portador, Sistema de inyección de muestras, Configuraciones de Columnas y Hornos, Sistemas de detección

51
Gases Portadores Deben ser químicamente inertes y presentar alta pureza. los de uso más común son Nitrógeno, Helio e Hidrógeno. La elección de dicho gas está relacionada con el tipo de detector que se utiliza. Con el suministro del gas se relacionan los reguladores de presión, los manómetros y los medidores de caudal. Además antes de la entrada del gas al instrumento, se le hace pasar por un tamiz molecular para eliminar la humedad y otras impurezas. El intervalo de presiones de entrada oscila comúnmente entre 10 y 50 psi. (sobre la presión circundante), lo que conduce a caudales de 25 a 150 mL/min en columnas empacadas y de 1 a 25 mL/min en columnas tubulares abiertas. El caudal se puede medir con instrumentos electrónicos, o con el caudalímetro de pompas de jabón.
, lo que conduce a caudales de 25 a 150 mL/min en columnas empacadas y de 1 a 25 mL/min en columnas tubulares abiertas. El caudal se puede medir con instrumentos electrónicos, o con el caudalímetro de pompas de jabón.")
52
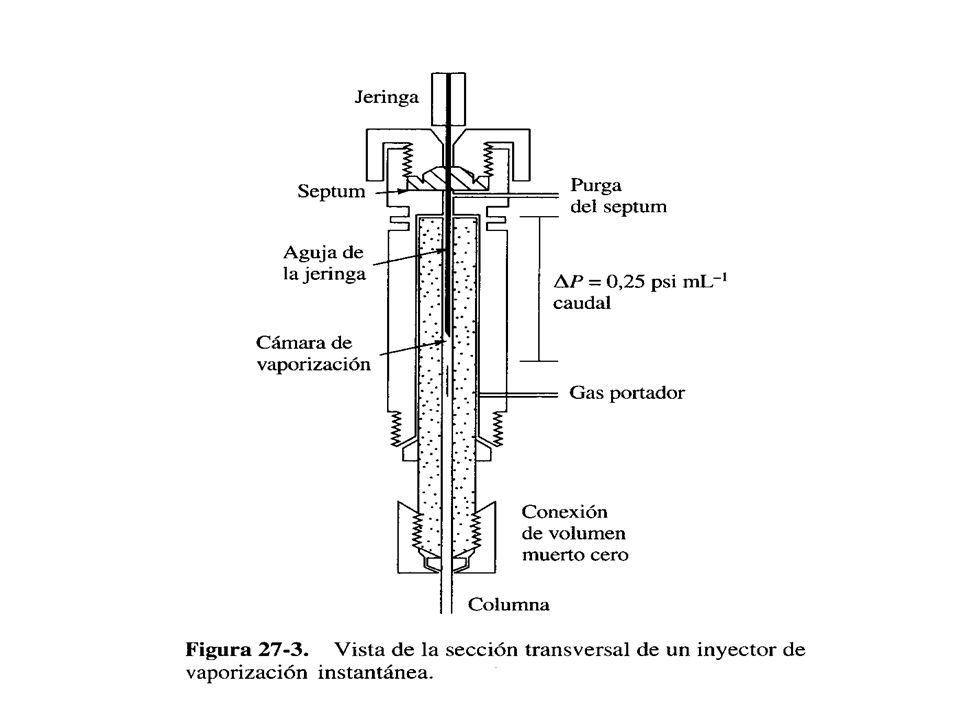
El cuál se coloca al final de la columna roscada sólo por la parte superior al inyector cuando se efectúa la medición del caudal. Sistema de inyección de muestra: La eficacia de la columna requiere que la muestra sea de un tamaño adecuado y que sea introducida como un tapón de vapor. La inyección lenta de muestras demasiado grandes provoca un ensanchamiento de bandas y una resolución muy pobre. El método más sencillo de inyectar la muestra es hacerlo manualmente, con una microjeringa calibrada a través de un diafragma o septum de goma de silicona en el puerto de inyección

53
Generalmente el puerto de inyección se mantiene a una temperatura de unos 50C por encima del punto de ebullición del componente menos volátil de la muestra. Los instrumentos más modernos están equipados con inyectores automáticos, los que para fines cuantitativos reducen notablemente el error experimental y permiten programar cantidades grandes de inyecciones y de muestras. El tamaño de la muestra varía desde menos de 1 L hasta 20 L para columnas empacadas, pero las columnas capilares exigen muestras mucho menores (~10³L). Suele ser necesario emplear un inyector divisor de muestras (Split-split less) para las columnas capilares con el fin de introducir una fracción pequeña conocida (1:100 a 1:500) de la muestra inyectada, mientras el resto se desecha por el canal de purga.
. Suele ser necesario emplear un inyector divisor de muestras (Split-split less) para las columnas capilares con el fin de introducir una fracción pequeña conocida (1:100 a 1:500) de la muestra inyectada, mientras el resto se desecha por el canal de purga..")
57
Configuraciones de columnas y hornos
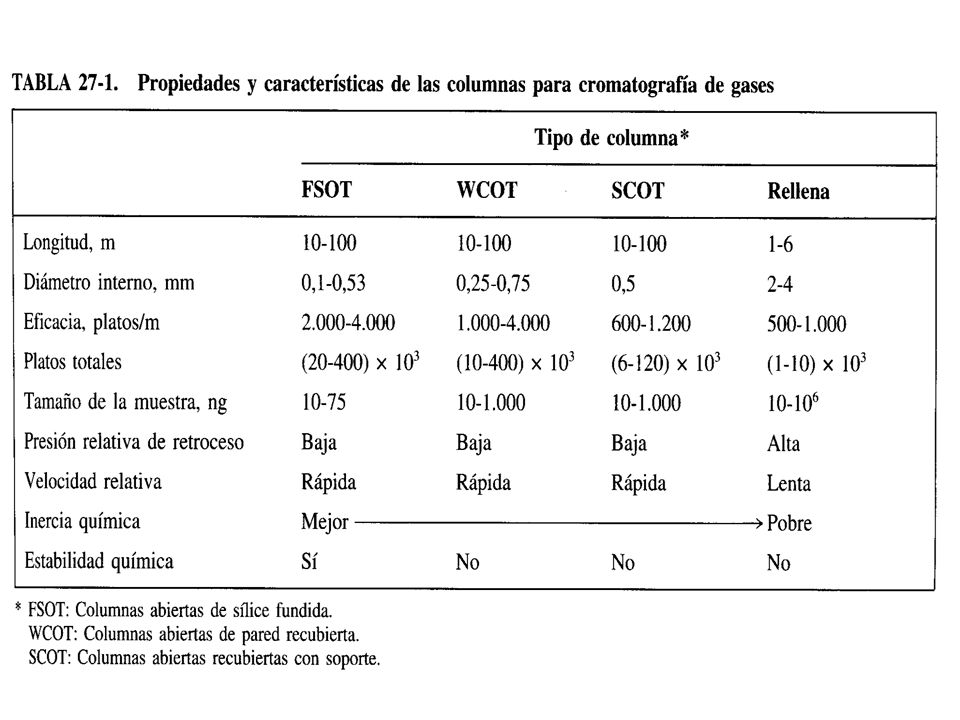
Los tipos generales de columnas en CGL son las columnas empacadas o rellenas y las columnas tubulares abiertas (capilares y megaboro). En el pasado la mayor parte de análisis por CGL se efectuaban con columnas empacadas, pero en la mayoría de las aplicaciones actuales han sido sustituidas por las columnas tubulares abiertas. La longitud de las columnas cromatográficas varía desde menos de 2 hasta más de 50 metros. Se han fabricado de acero inoxidable, vidrio, sílice fundida y teflón.
. En el pasado la mayor parte de análisis por CGL se efectuaban con columnas empacadas, pero en la mayoría de las aplicaciones actuales han sido sustituidas por las columnas tubulares abiertas. La longitud de las columnas cromatográficas varía desde menos de 2 hasta más de 50 metros. Se han fabricado de acero inoxidable, vidrio, sílice fundida y teflón.")
58
Corte transversal de columnas tubulares abiertas

60
Para lograr que las columnas encajen dentro del horno para su calentamiento, es habitual enrollarlas en serpentines con diámetros entre 10 y 30 cm. La temperatura de las columnas es una variable importante y para trabajar con precisión debe controlarse con una exactitud de décimas de grado. Generalmente una temperatura igual o un poco mayor que el punto de ebullición promedio de las muestras permite una duración razonable de análisis (2-30 min). En el caso de muestras con un intervalo de ebullición amplio, es necesario utilizar una programación de temperaturas, con la que se incremente la temperatura de la columna de forma continua o escalonada, a medida que avanza la separación.
. En el caso de muestras con un intervalo de ebullición amplio, es necesario utilizar una programación de temperaturas, con la que se incremente la temperatura de la columna de forma continua o escalonada, a medida que avanza la separación..")
61
Ejemplo del efecto de la temperatura sobre los cromatogramas

62
Sistemas de detección Se han investigado y utilizado docenas de detectores durante el desarrollo de la CGL, por lo que se estudiarán aquellos de uso más frecuente y los más poderosos. Dado que el detector ideal debe poseer Sensibilidad adecuada Buena estabilidad y reproducibilidad Respuesta lineal para los solutos extendida a varias órdenes de magnitud Intervalo de temperaturas de ambiente hasta 400C Tiempo de respuesta corto independiente del caudal Alta confiabilidad y manejo sencillo Respuesta semejante para todos los solutos No destructivo de la muestra

63
Hasta la fecha, aún no se ha diseñado un detector que reúna todas las cualidades mencionadas, por lo que se mencionarán las características de los de uso más común: Detector de ionización de llama (FID) que es el que ha sido más ampliamente utilizado Detector de Captura de Electrones (ECD), específico para detectar y cuantificar compuestos halogenados Detector de Conductividad Térmica ó Catarómetro, que fue el primer detector utilizado, y Detector de Masas (MS) que por el momento es el más avanzado.
 que es el que ha sido más ampliamente utilizado. Detector de Captura de Electrones (ECD), específico para detectar y cuantificar compuestos halogenados. Detector de Conductividad Térmica ó Catarómetro, que fue el primer detector utilizado, y. Detector de Masas (MS) que por el momento es el más avanzado.")
64
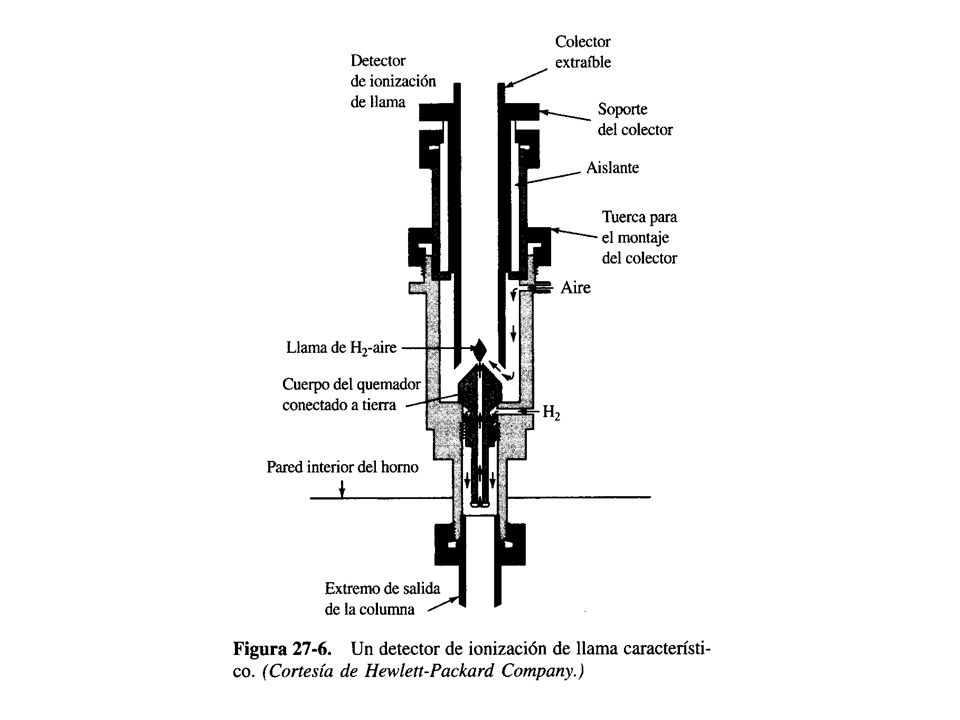
DETECTOR DE IONIZACIÓN POR LLAMA
Este tipo de detector mide la corriente que puede pasar entre un par de electrodos con polarización opuesta colocados a ambos lados de una llama de hidrógeno/aire. La llama produce un plasma gaseoso que tiene una alta resistividad eléctrica en ausencia de iones. La alta temperatura de la llama piroliza la mayor parte de los compuestos orgánicos y produce cationes y electrones, que funcionan como portadores de carga entre los dos electrodos. Los iones se recolectan en el ánodo llamado colector. La corriente que pasa se puede amplificar y los componentes eluidos de la columna se registran como picos cromatográficos. La respuesta depende de la cantidad de átomos de carbono de la molécula del analito y también, de su concentración.

65
También el estado de oxidación/reducción del carbono puede afectar hasta cierto grado la sensibilidad de este detector y el carbono totalmente oxidado no se ioniza dentro de la llama. El gas de arrastre normal puede ser HELIO, ARGÓN o NITRÓGENO, debido a sus estabilidades térmicas, no combustibilidad e inercia química. El detector de ionización es el de uso más generalizado debido a su simplicidad, robustez y alta sensibilidad, que puede permitir determinaciones hasta de 10¹³ g/cm³, así como a su capacidad para responder dentro de un margen amplio de concentraciones. La desventaja mayor que presenta el detector FID es que la llama destruye la muestra al pirolizarla para formar los iones corrrespondientes.

67
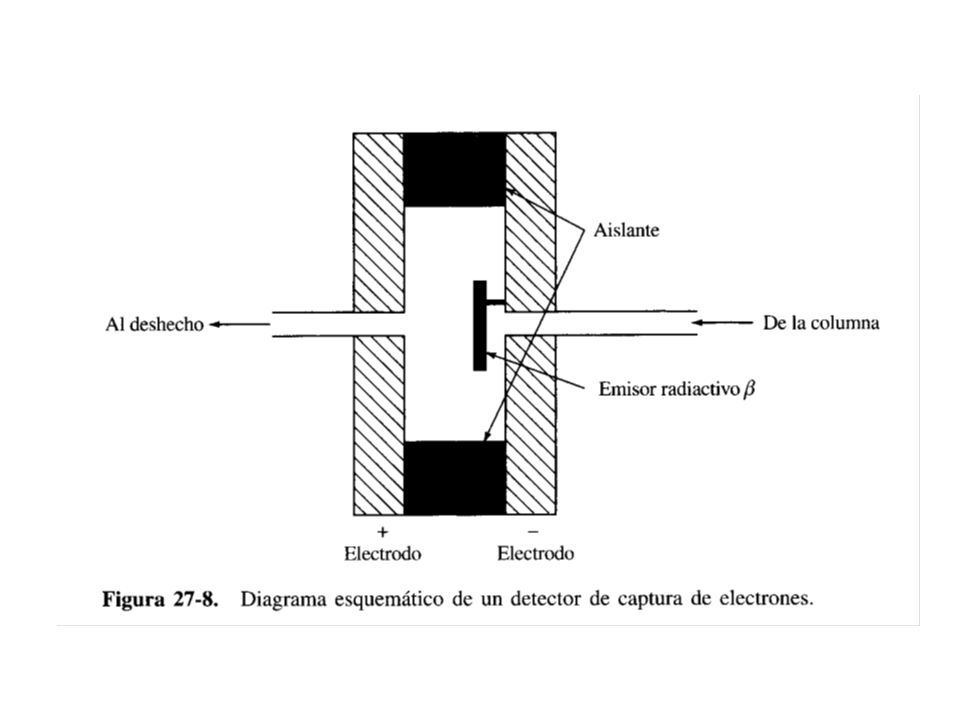
DETECTOR DE CAPTURA DE ELECTRONES
En este tipo de detector, para irradiar el eluido que sale de la columna, se usa un emisor de partículas como 63Ni o Tritio. Las partículas (electrones) causan ionización del gas de arrastre (por ejemplo N2) y se produce más liberación de electrones. El detector posee un par de electrodos de polarización opuesta colocados en ambos lados de la corriente del gas eluido y al medir la corriente que pasa entre los electrodos, se produce la respuesta del detector. Los compuestos orgánicos con tendencia a capturar electrones, al capturar a los mismos, producen una disminución de la corriente entre los electrodos, por lo que una disminución de corriente corresponderá a un pico cromatográfico al eluirse un analito de la columna.
 causan ionización del gas de arrastre (por ejemplo N2) y se produce más liberación de electrones. El detector posee un par de electrodos de polarización opuesta colocados en ambos lados de la corriente del gas eluido y al medir la corriente que pasa entre los electrodos, se produce la respuesta del detector. Los compuestos orgánicos con tendencia a capturar electrones, al capturar a los mismos, producen una disminución de la corriente entre los electrodos, por lo que una disminución de corriente corresponderá a un pico cromatográfico al eluirse un analito de la columna.")
69
CROMATOGRAFÍA DE GASES ACOPLADA A OTROS INSTRUMENTOS DETECTORES
Con frecuencia, la CG y CGL se acopla con otras técnicas analíticas para ampliar la sensibilidad. Con dichos métodos se aprovecha la capacidad de separación de la cromatografía para fraccionar a la muestra en sus partes componentes y luego permitir la cuantificación mediante por ejemplo, Espectroscopía de Masas (GC-MS), Espectroscopía Infrarroja (GC-IR), ó Espectroscopía de Resonancia Magnética Nuclear (GC-NMR).
, Espectroscopía Infrarroja (GC-IR), ó Espectroscopía de Resonancia Magnética Nuclear (GC-NMR).")
71
CROMATOGRAFÍA DE LÍQUIDOS

73
CROMATOGRAFIA DE LÍQUIDOS
En la primeras etapas de desarrollo de la CL, los científicos se dieron cuenta que se podían conseguir grandes aumentos en la eficacia de la columna al disminuir el tamaño de las partículas de los rellenos. Hasta finales de la década de los años 70s, se logró diseñar la tecnología adecuada para producir y utilizar rellenos de partícula tan pequeña, como los del orden de 3 a 10m. Esta tecnología requiere de un instrumental sofisticado para poder trabajar a altas presiones, lo cuál contrasta notablemente con las sencillas columnas de vidrio de la cromatografía de líquidos clásica cuyo caudal se debe a la gravedad. Para distinguir este procedimiento tan sofisticado de los métodos básicos que todavía pueden emplearse para fines preparativos, se usa la denominación de Cromatografía de Líquidos de Alta Eficacia (HPLC).
.")
74
HPLC es la técnica de separación más ampliamente utilizada, debido a su sensibilidad, su fácil adaptación a las determinaciones cuantitativas exactas, su idoneidad para la separación de especies no volátiles o termolábiles y principalmente a su gran aplicabilidad a sustancias que son de primordial interés en la industria, en muchos campos de la ciencia y para la sociedad en general. Entre estos materiales se incluyen: Aminoácidos, proteínas, ácidos nucléicos, hidrocarburos, carbohidratos, fármacos, terpenoides, plaguicidas, antibióticos, esteroides, especies organometálicas y una variedad de sustancias inorgánicas.

76
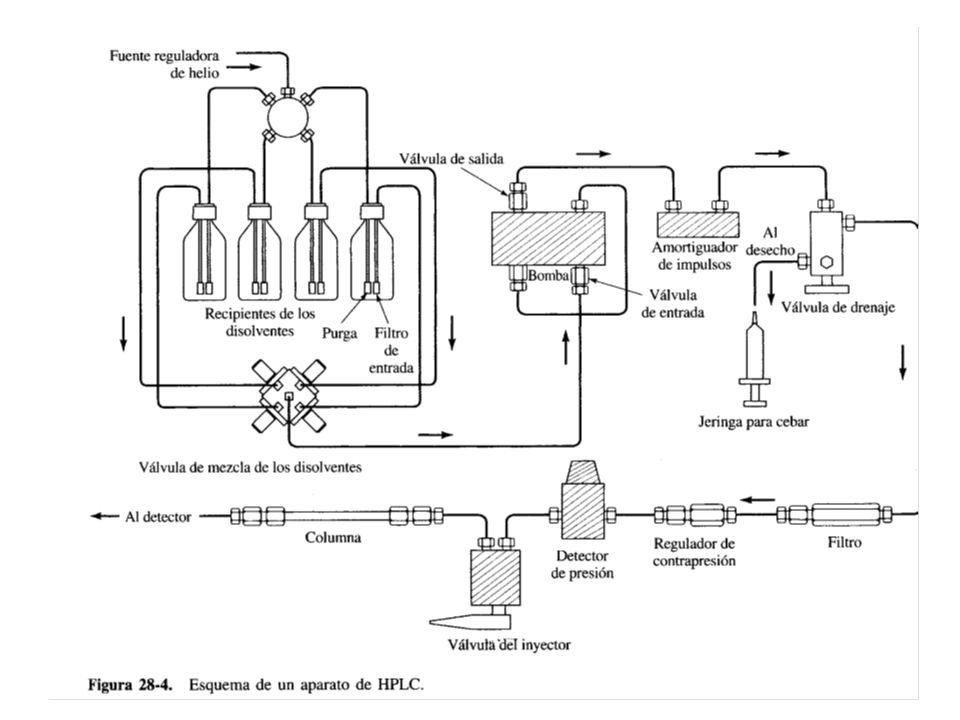
INSTRUMENTACIÓN para HPLC: Un equipo moderno de HPLC, está equipado con dos o más recipientes de vidrio que pueden contener hasta 1000 mL de disolvente. El instrumento puede incluir un sistema para eliminar los gases disueltos (oxígeno y nitrógeno, generalmente) que al formar burbujas interfieren en los sistemas de detección, provocando ensanchamiento de banda e interfieren todo el funcionamiento del detector. Este desgasificador puede consistir en un sistema de bombeo por vacío, o por agitación y también de sistemas de purga para arrastrar los gases disueltos fuera de la solución. Estos sistemas también pueden contener un dispositivo para filtrar el polvo y otras partículas sólidas suspendidas en los disolventes, pues ellas pueden dañar la bomba o los sistemas de inyección u obturar la columna.
 que al formar burbujas interfieren en los sistemas de detección, provocando ensanchamiento de banda e interfieren todo el funcionamiento del detector. Este desgasificador puede consistir en un sistema de bombeo por vacío, o por agitación y también de sistemas de purga para arrastrar los gases disueltos fuera de la solución. Estos sistemas también pueden contener un dispositivo para filtrar el polvo y otras partículas sólidas suspendidas en los disolventes, pues ellas pueden dañar la bomba o los sistemas de inyección u obturar la columna..")
77
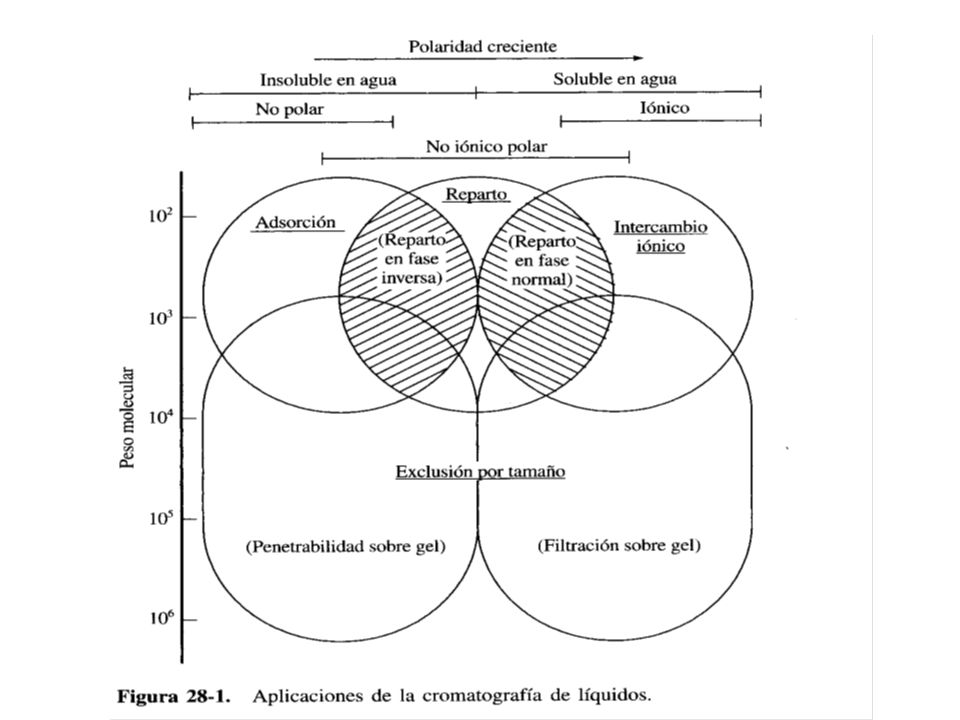
Cromatografía de reparto (o de partición)
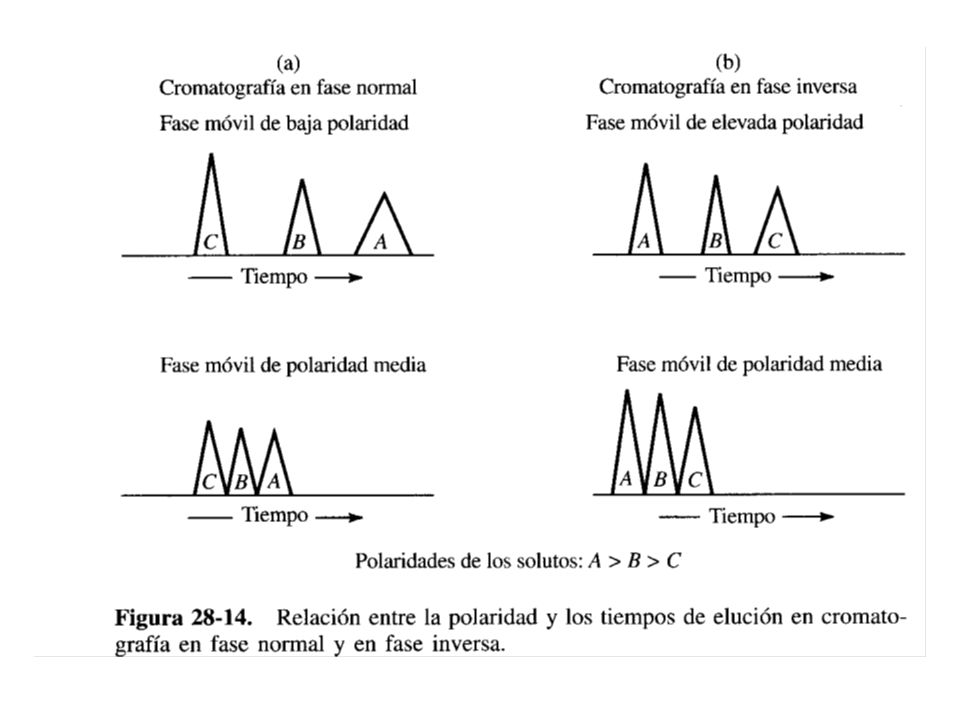
Este tipo de cromatografía ha llegado a ser el más ampliamente utilizado de los cuatro tipos de cromatografía de líquidos (adsorción, reparto o partición, intercambio iónico y exclusión por tamaño). En relación con las polaridades de las fases móvil y estacionaria, se distinguen dos tipos de cromatografía de reparto: Al principio, en CL se utilizaban fE de elevada polaridad tales como el agua y el trietilenglicol soportada sobre partículas de sílice o alúmina y como fase móvil se empleaba un disolvente relativamente apolar como hexano o isopropileter. Por razones históricas, a este tipo de cromatografía se le conoce actualmente como Cromatografía en fase Normal.
. En relación con las polaridades de las fases móvil y estacionaria, se distinguen dos tipos de cromatografía de reparto: Al principio, en CL se utilizaban fE de elevada polaridad tales como el agua y el trietilenglicol soportada sobre partículas de sílice o alúmina y como fase móvil se empleaba un disolvente relativamente apolar como hexano o isopropileter. Por razones históricas, a este tipo de cromatografía se le conoce actualmente como Cromatografía en fase Normal.")
78
En la cromatografía de fase reversa, la fE es apolar (generalmente un hidrocarburo) y la fM es polar (agua, metanol, acetonitrilo). Aproximadamente el 75% de toda la cromatografía de líquidos de alta eficiencia se efectúan en columnas con rellenos de fase reversa. Sistemas de bombeo La muestra que contiene a los analitos se disuelve en un disolvente o en una mezcla de disolventes. Se selecciona la fM para permitir la separación más eficiente en un tiempo mínimo. Se suele escoger la fM como una mezcla parecida a la que disolvió a la muestra, por lo que la fM también puede contener uno o más disolventes para facilitar la separación.

79
La muestra se inyecta en el flujo de la fase móvil e ingresan, a presión, a la columna mediante el uso de una bomba. Los requisitos para un sistema de bombeo en HPLC son rigurosos, e incluyen, entre otros, la resistencia a la generación de presiones por encima de 6000 psi. Existen varios tipos de bombas para HPLC, siendo la bomba Recíproca la que se usa en el 90% de los sistemas de HPLC actuales.

80
Sistema de inyección de la muestra
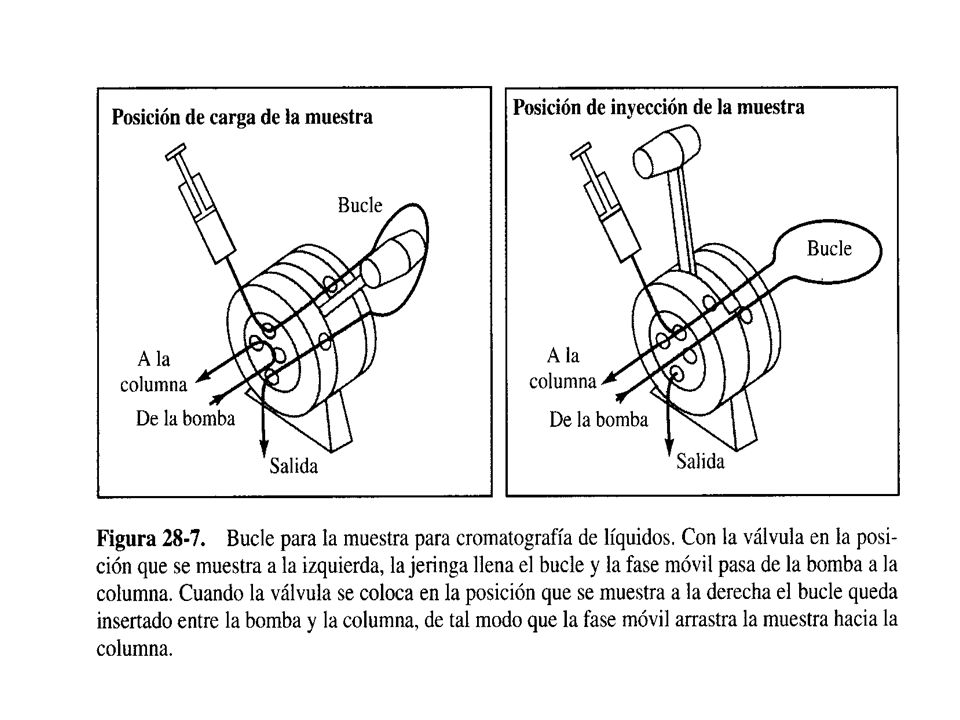
El factor limitante en la precisión de la mediciones en HPLC es la reproducibilidad con que se pueda introducir la muestra en la columna. Los volúmenes que se empleen han de ser pequeños, desde unas décimas de L hasta 500 L . Es muy importante introducir la muestra sin despresurizar el sistema. El método más ampliamente utilizado para ese propósito es usar Bucles de Muestra (Loops) que normalmente son parte integral del instrumento. Con ellos se pueden inyectar volúmenes definidos y que también puede modificarse con el software. Con este tipo de dispositivos se puede introducir la muestra con una precisión relativa de unas décimas por ciento.
 que normalmente son parte integral del instrumento. Con ellos se pueden inyectar volúmenes definidos y que también puede modificarse con el software. Con este tipo de dispositivos se puede introducir la muestra con una precisión relativa de unas décimas por ciento.")
82
Columnas para HPLC Comúnmente las columnas para HPLC se empacan dentro de tubos de acero inoxidable para resistir las altas presiones que se usen. Los diámetros internos típicos del tubo pueden ser entre 3 y 10 mm. Y sus longitudes entre 10 y 30 cms. Los diámetros de partículas van de 3 a 10 m, para dar (teóricamente) de 40,000 a 100,000 platos por metro. Materiales de empaque: Los de uso más común son Pelicular y de Partícula Porosa. Las partículas de empaque pelicular se preparan con perlas de vidrio o de polímero no poroso, de forma esférica y con diámetros entre 30 y 40 m. Sobre la superficie de las perlas se depositan capas porosas
 de 40,000 a 100,000 platos por metro. Materiales de empaque: Los de uso más común son Pelicular y de Partícula Porosa. Las partículas de empaque pelicular se preparan con perlas de vidrio o de polímero no poroso, de forma esférica y con diámetros entre 30 y 40 m. Sobre la superficie de las perlas se depositan capas porosas.")
83
Y delgadas de por ejemplo, una resina de intercambio iónico, sílice o alúmina, o también una resina sintética de poliestireno-divinilbenceno. Por lo general los rellenos peliculares se utilizan ampliamente en las precolumnas y no en las columnas analíticas. Los rellenos típicos de partículas porosas para HPLC, están formados por micro partículas porosas con diámetros entre 3 y 10 m. y con la menor dispersión posible con respecto de un tamaño determinado. Es muy importante mantener el diámetro de partícula tan uniforme como sea posible, porque es uno de los factores que afecta de modo crucial la reproducibilidad de la eficiencia de separación en las columnas empacadas. El material de las partículas puede ser sílice, alúmina o de resinas de intercambio iónico y/o de poliestireno-divinilbenceno, siendo sílice el material más

84
Ampliamente utilizado.
Las partículas de sílice se preparan aglomerando partículas de sílice de tamaños inferiores a 1 m, bajo condiciones tales que permiten que se formen partículas mayores con diámetros muy uniformes. Las partículas que resultan pueden recubrirse también con películas orgánicas que se unen química o físicamente a la superficie. En cromatografía de reparto los soportes para la mayoría de rellenos de fase químicamente enlazada se preparan con sílice rígida o composiciones constituidas básicamente por sílice. Estos sólidos están formados por partículas mecánicamente resistentes, porosas y uniformes, con diámetros de 3.5 ó 10 m. La superficie de sílice que ha sido totalmente hidrolizada por medio de calentamiento

85
Con HCl 0.1 M, durante uno o dos días, está constituida por grupos silanol químicamente reactivos. Es decir, que las superficies de sílice características contienen cerca de 8 m/m² de grupos OH. Los recubrimientos de fase unida químicamente más utilizados son los siloxanos, que se forman por reacción de la superficie hidrolizada con un silano organoclorado,

86
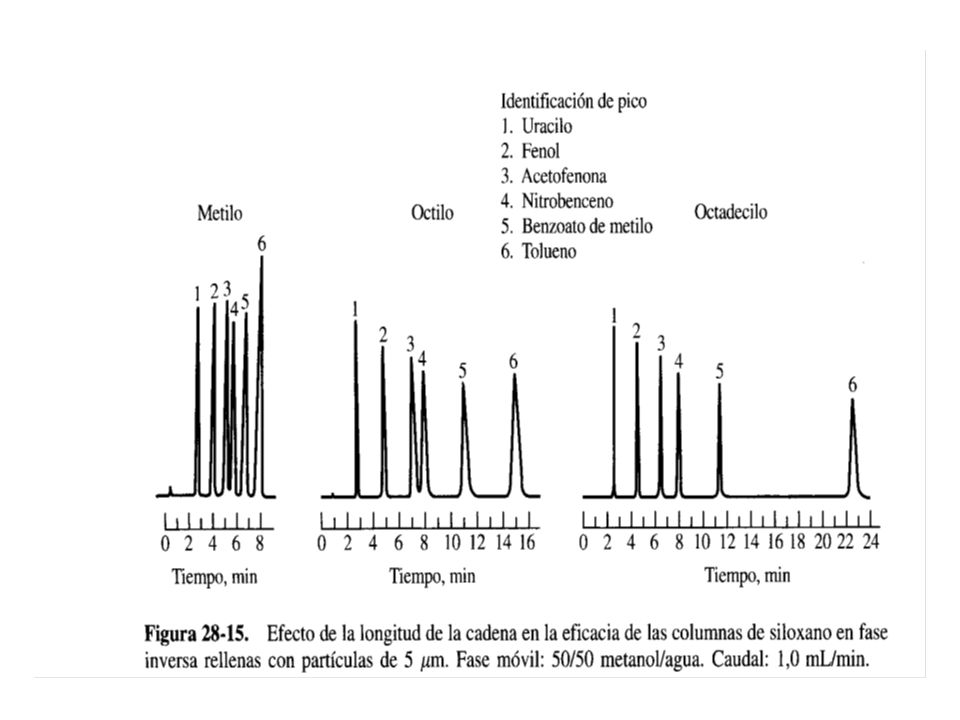
Por ejemplo: Donde R es un grupo alquilo o alquilo sustituido. Por lo común, el grupo R del siloxano en estos recubrimientos es una cadena C8 (n-octilo) o una cadena C18 (n-octadecilo). En estas fases, los grupos de hidrocarburo de cadena larga se alinean uno junto al otro y en perpendicular a la superficie de la partícula, presentando una estructura semejante a una brocha o a un cepillo. Hasta el momento no se ha establecido el mecanismo por el cuál esta estructura retiene a los solutos.
 o una cadena C18 (n-octadecilo). En estas fases, los grupos de hidrocarburo de cadena larga se alinean uno junto al otro y en perpendicular a la superficie de la partícula, presentando una estructura semejante a una brocha o a un cepillo. Hasta el momento no se ha establecido el mecanismo por el cuál esta estructura retiene a los solutos.")
88
En relación con las polaridades de las fases móvil y estacionaria, se distinguen dos tipos de cromatografía de reparto: Al principio, en CL se utilizaban fE de elevada polaridad tales como el agua y el trietilenglicol soportada sobre partículas de sílice o alúmina y como fase móvil se empleaba un disolvente relativamente apolar como hexano o isopropileter. Por razones históricas, a este tipo de cromatografía se le conoce actualmente como Cromatografía en fase Normal. En la Cromatografía de fase Reversa, la fE es apolar (generalmente un hidrocarburo) y la fM es polar (agua, metanol, acetonitrilo). Aproximadamente el 75% de toda la cromatografía de líquidos de alta eficiencia se efectúan en columnas con rellenos de fase reversa.
 y la fM es polar (agua, metanol, acetonitrilo). Aproximadamente el 75% de toda la cromatografía de líquidos de alta eficiencia se efectúan en columnas con rellenos de fase reversa.")
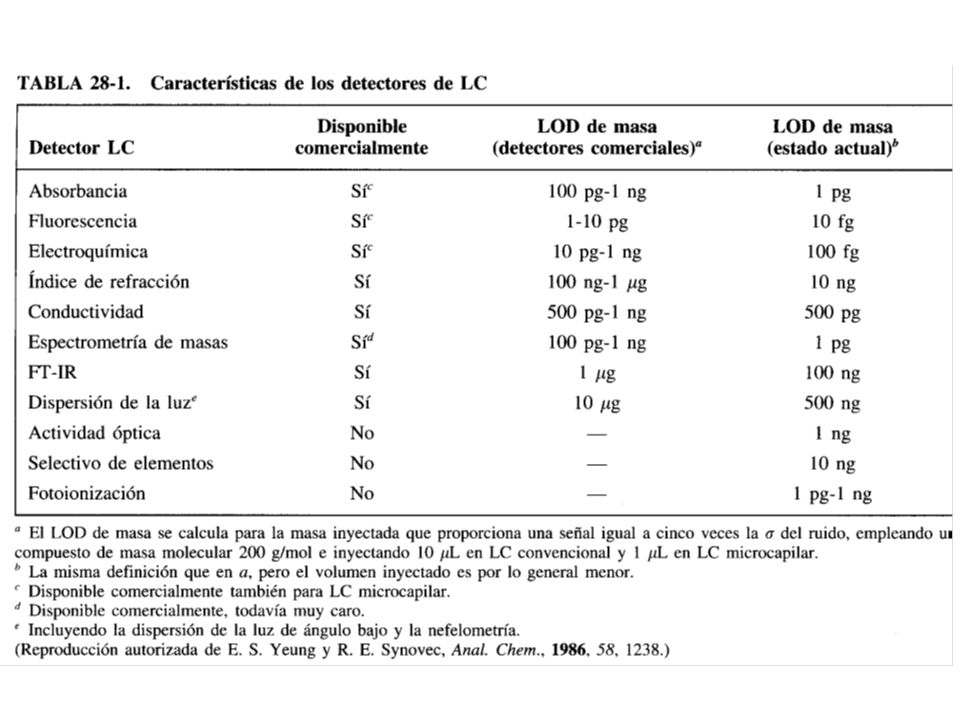
90
DETECTORES A diferencia de CG, en HPLC no existen detectores tan aplicables a todo tipo de muestras. Un detector para HPLC debería tener las mismas cualidades mencionadas para los de CG, excepto ser sensible en un intervalo grande de temperaturas, pero si poseer un volumen mínimo interno para reducir el ensanchamiento de banda. Los detectores para HPLC son de dos tipos básicos, a. Los que se basan en la medición de una propiedad de la disolución respondiendo a una propiedad del efluente, tal como el índice de refracción, la constante dieléctrica o la densidad que se modifica por la presencia del analito.

91
Por el contrario, b. los detectores que se basan en una propiedad del soluto o muestra, como la absorbancia en UV, Fluorescencia o corriente límite, que no son inherentes a la fase móvil. Detectores de absorbancia Ultra Violeta Pueden contener dispositivos de doble haz, de un solo haz, filtros o monocromadores, pero los más potentes son los que están equipados con diodos en serie.

93
Detección por Espectrometría de Masas
La Espectrometría de Masas se usa como una poderosa herramienta de detección acoplada a HPLC, ya que se puede identificar la masa de cada especie molecular a medida que se eluye de la columna. El problema fundamental de acoplar la HPLC con la Espectrometría de Masas es que la HPLC utiliza cantidades grandes (relativamente) de disolvente en la fase móvil, mientras que en la Espectrometría de Masas se requiere que la muestra se introduzca en una cámara a alto vacío. Para resolver dicho problema se han desarrollado varias interfases, siendo la ESI (Electro Spray Ionization) una de las más utilizadas, en ella, el efluente de la columna cromatográfica pasa por un tubo capilar de acero inoxidable calentado, para evaporar gran parte del disolvente, formado un aerosol de analito y disolvente.
 de disolvente en la fase móvil, mientras que en la Espectrometría de Masas se requiere que la muestra se introduzca en una cámara a alto vacío. Para resolver dicho problema se han desarrollado varias interfases, siendo la ESI (Electro Spray Ionization) una de las más utilizadas, en ella, el efluente de la columna cromatográfica pasa por un tubo capilar de acero inoxidable calentado, para evaporar gran parte del disolvente, formado un aerosol de analito y disolvente.")
94
El acoplamiento HPLC-MS puede ofrecer grados extremadamente altos de sensibilidad y límites de detección diminutos.

95
El establecimiento del método en cromatografía de reparto tiende a ser más complejo que en CG, debido a que cuando la fM es líquida los componentes de la muestra interaccionan con ambas fases, la fE y la fM. Una cromatografía exitosa con fases móviles interactivas requiere un equilibrio adecuado entre las fuerzas intermoleculares existentes entre los tres participantes activos en el proceso de la separación (El soluto, la fase móvil y la fase estacionaria). Estas fuerzas intermoleculares se describen cualitativamente en términos de polaridad relativa de cada uno de los tres participantes. Las polaridades en orden creciente para varios grupos funcionales de los analitos son : Hidrocarburos <éteres <ésteres <cetonas < aldehídos <amidas <aminas <alcoholes. El agua es más polar que cualquier compuesto que contenga alguno de estos grupos.
. Estas fuerzas intermoleculares se describen cualitativamente en términos de polaridad relativa de cada uno de los tres participantes. Las polaridades en orden creciente para varios grupos funcionales de los analitos son : Hidrocarburos <éteres <ésteres <cetonas < aldehídos <amidas <aminas <alcoholes. El agua es más polar que cualquier compuesto que contenga alguno de estos grupos.")
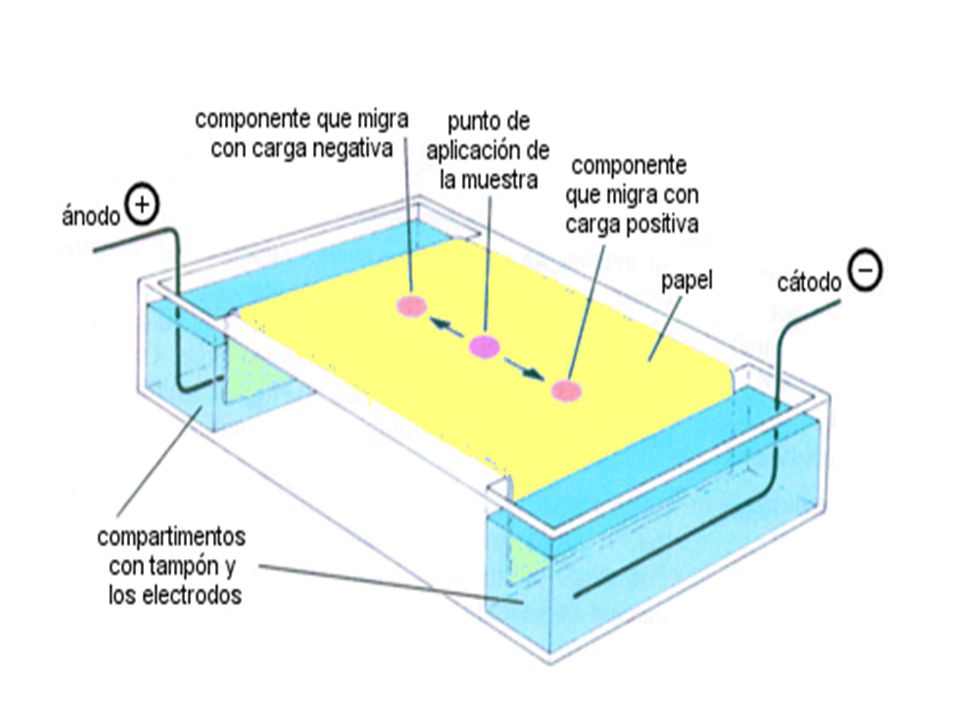
97
ELECTROFORESIS Con la técnica de electroforesis se pueden separar sustancias por MIGRACIÓN ELÉCTRICA, de modo que las sustancias ionizadas, o cuando menos muy polares, se mueven bajo la influencia de un campo eléctrico. La rapidez de movimiento de una sustancia es función tanto de su peso molecular como de su carga debida a la resistencia al movimiento, esto es, la resistencia viscosa que presenta el medio a través del cuál debe moverse. Existen muchas formas de electroforesis, una de las más versátiles y probablemente la que se usa con más frecuencia se llama Electroforesis de Zona, en la que los solutos se mueven dentro de una fase móvil a través de un soporte estacionario. Por consiguiente los análisis de este tipo aprovechan los métodos cromatográficos

98
Y los de migración para lograr la separación que se requiera
Y los de migración para lograr la separación que se requiera. Comúnmente las fases estacionarias de soporte se empacan o fabrican en forma de un bloque o de una placa, con almidón, gel de poliacrilamida, espuma de poliuretano, agarosa o hasta con papel. La electroforesis en gel de almidón o de poliacrilamida consiste en depositar una banda angosta de la mezcla de muestra cruzando una línea a la mitad de los lados del bloque de material. Los dos extremos del bloque se conectan con electrodos a una fuente de alto potencial y al polarizar el bloque los distintos componentes (solutos ionizados) migran hacia el ánodo o hacia el cátodo, dependiendo de sus polaridades respectivas. Las bandas o líneas separadas corresponden a solutos en la mezcla y corrientemente
 migran hacia el ánodo o hacia el cátodo, dependiendo de sus polaridades respectivas. Las bandas o líneas separadas corresponden a solutos en la mezcla y corrientemente.")
99
Se revelan tiñendo los componentes después de la separación
Se revelan tiñendo los componentes después de la separación. A continuación se puede usar un densitómetro para determinar las intensidades de las bandas coloreadas y en esta forma se pueden cuantificar las concentraciones relativas de diversos componentes en la mezcla, por referencia a un perfil predeterminado de calibración de intensidades de color. El campo eléctrico a través de la placa o bloque se expresa en términos de voltios (volts) por centímetro y sus valores pueden ser de 500 a 5000 V cm-¹ o mayores. Los solutos de mayores pesos moleculares (como las macromoléculas de proteínas) requieren con frecuencia mayores campos eléctricos para la separación, que los solutos de peso molecular menor. Se debe tomar en cuenta que el pH afecta el grado de ionización de los solutos en la mezcla y por consiguiente
 por centímetro y sus valores pueden ser de 500 a 5000 V cm-¹ o mayores. Los solutos de mayores pesos moleculares (como las macromoléculas de proteínas) requieren con frecuencia mayores campos eléctricos para la separación, que los solutos de peso molecular menor. Se debe tomar en cuenta que el pH afecta el grado de ionización de los solutos en la mezcla y por consiguiente.")
100
La rapidez y el grado de separación
La rapidez y el grado de separación. Esto tiene relevancia especial cuando se analiza el contenido de aminoácidos en mezclas, que con frecuencia se separan en los laboratorios clínicos o bioquímicos con Electroforesis de Zona. A cierto pH (el llamado punto isoeléctrico), la carga neta que porta determinado aminoácido será cero, ya que existirá en forma de zwiteriones, bajo dichas condiciones el aminoácido no se moverá ni hacia el ánodo ni hacia el cátodo. La electroforesis convencional se ha aplicado a un conjunto de separaciones analíticas dificultosas: aniones y cationes inorgánicos, aminoácidos, catecolaminas, fármacos, vitaminas, carbohidratos, péptidos, proteínas, ácidos nucleicos, nucleótidos, polinucleótidos y otras especies.
, la carga neta que porta determinado aminoácido será cero, ya que existirá en forma de zwiteriones, bajo dichas condiciones el aminoácido no se moverá ni hacia el ánodo ni hacia el cátodo. La electroforesis convencional se ha aplicado a un conjunto de separaciones analíticas dificultosas: aniones y cationes inorgánicos, aminoácidos, catecolaminas, fármacos, vitaminas, carbohidratos, péptidos, proteínas, ácidos nucleicos, nucleótidos, polinucleótidos y otras especies.")
103
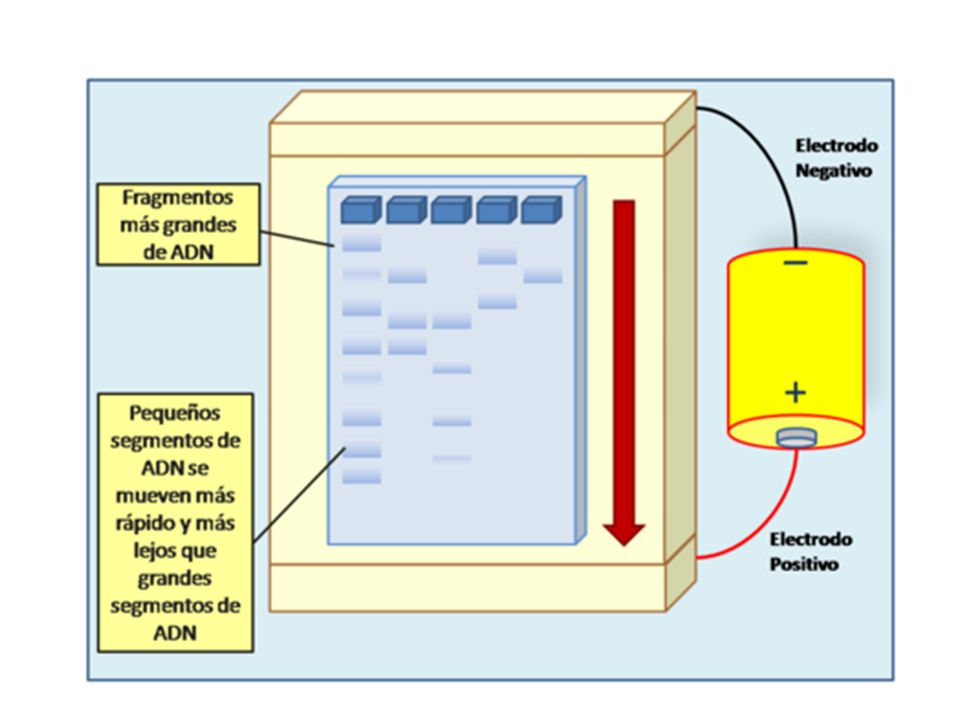
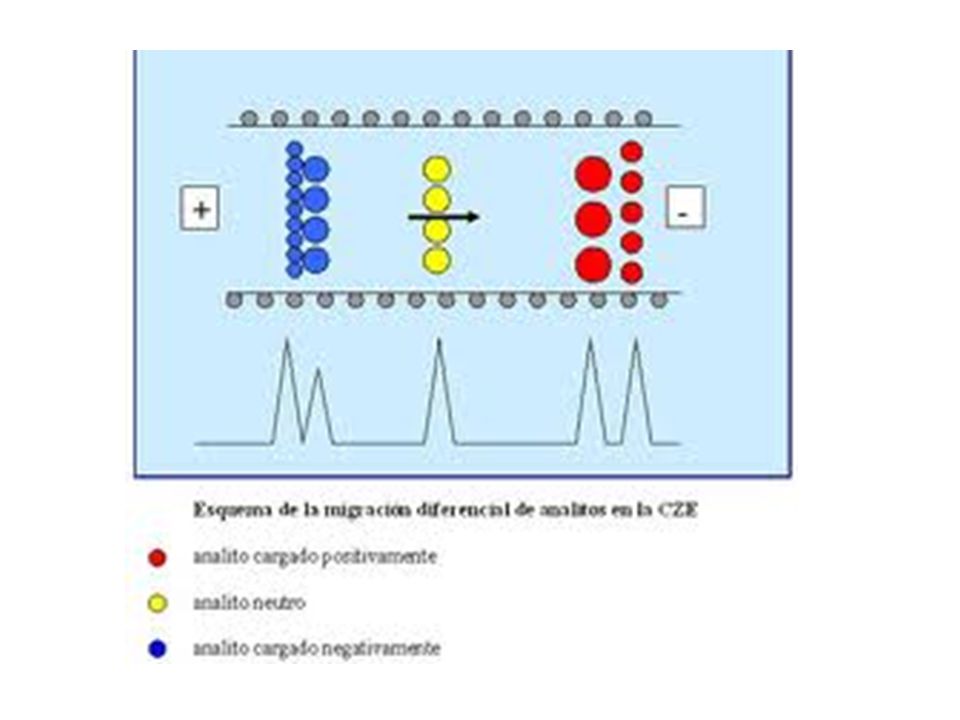
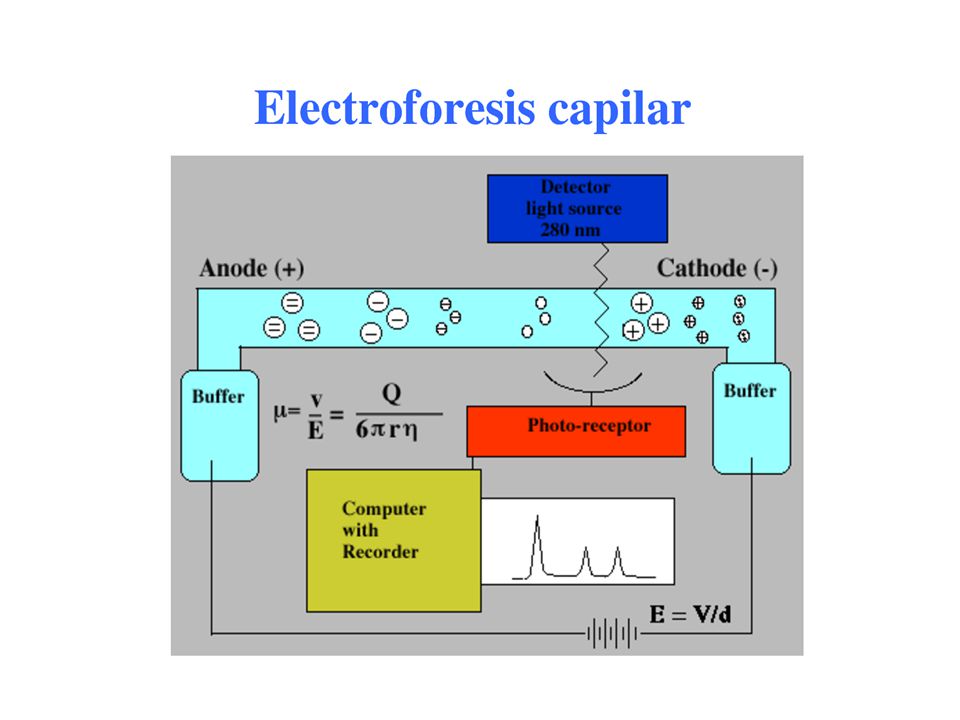
ELECTROFORESIS CAPILAR
La electroforesis capilar de zona se está usando cada vez más en los laboratorios biológicos, pues con volúmenes pequeños de muestra (del orden de 1 nL) de muestras complicadas, se pueden separar y cuantificar sus componentes con relativa facilidad. La instrumentación incluye tubos capilares de sílice fundida, típicamente de 1 m de longitud y de entre 10 y 100 m de diámetro interno. Dicho capilar se llena con el electrolito de elección. A continuación se introduce un pequeño volumen de la muestra a separar por un extremo del tubo, ya sea por simple inmersión en la mezcla o mediante entrada hidrostática o neumática. En algunos casos se puede introducir la muestra aplicando un pequeño potencial a través de los extremos del capilar, para inducir una succión de
 de muestras complicadas, se pueden separar y cuantificar sus componentes con relativa facilidad. La instrumentación incluye tubos capilares de sílice fundida, típicamente de 1 m de longitud y de entre 10 y 100 m de diámetro interno. Dicho capilar se llena con el electrolito de elección. A continuación se introduce un pequeño volumen de la muestra a separar por un extremo del tubo, ya sea por simple inmersión en la mezcla o mediante entrada hidrostática o neumática. En algunos casos se puede introducir la muestra aplicando un pequeño potencial a través de los extremos del capilar, para inducir una succión de.")
104
Los solutos por migración eléctrica
Los solutos por migración eléctrica. Los extremos del tubo capilar se sumergen entonces en dos recipientes separados, de solución buffer en la que están inmersos electrodos de platino. Con frecuencia se usa tubo capilar que contenga grupos silanol ionizables, que se cargarán negativamente en ambientes de pH 2 o mayores. Los cationes (iones con carga positiva) se adsorben a lo largo de la pared interna del capilar y forman una capa doble o contraria de carga. A través del capilar se aplica un potencial de 1,000 a 30,000 V o más, lo cuál hace que los aniones (iones con carga negativa) migren hacia el cátodo (electrodo negativo) bajo la influencia de las migraciones eléctricas. Dado que esos iones están solvatados arrastran moléculas de solvente y de todos los demás solutos en la misma dirección causando un flujo de fluido unidireccional (electroósmosis) hacia el cátodo. Las velocidades de flujo osmótico suelen ser del
 se adsorben a lo largo de la pared interna del capilar y forman una capa doble o contraria de carga. A través del capilar se aplica un potencial de 1,000 a 30,000 V o más, lo cuál hace que los aniones (iones con carga negativa) migren hacia el cátodo (electrodo negativo) bajo la influencia de las migraciones eléctricas. Dado que esos iones están solvatados arrastran moléculas de solvente y de todos los demás solutos en la misma dirección causando un flujo de fluido unidireccional (electroósmosis) hacia el cátodo. Las velocidades de flujo osmótico suelen ser del.")
105
Orden de varios cientos de nanolitros por minuto, aunque esto depende del pH del electrolito, del potencial aplicado y de la concentración de la solución buffer. Los analitos con carga más positiva (catiónicos) y con los pesos moleculares más bajos son los que se detectarán primero. Las moléculas neutras suelen moverse a un flujo un poco menor que el flujo osmótico, ya que no las influencia el campo eléctrico en forma directa, sino sólo el flujo del disolvente. Los aniones con carga más negativa viajarán con el flujo mínimo y en consecuencia serán detectados por último, pues el campo eléctrico retardará su movimiento.
 y con los pesos moleculares más bajos son los que se detectarán primero. Las moléculas neutras suelen moverse a un flujo un poco menor que el flujo osmótico, ya que no las influencia el campo eléctrico en forma directa, sino sólo el flujo del disolvente. Los aniones con carga más negativa viajarán con el flujo mínimo y en consecuencia serán detectados por último, pues el campo eléctrico retardará su movimiento.")
110
Existen varias modalidades de Electroforesis Capilar, entre ellas: Electroforesis capilar de zona, Electroforesis capilar en Gel, Isoelectroenfoque Capilar e Isotacoforesis Capilar. Debido a que en la mayoría de las modalidades de electroforesis capilar los analitos separado se desplazan pasando por un punto común, los detectores son semejantes en diseño y en función a los de HPLC.

Presentaciones similares














>")











