Descargar la presentación
La descarga está en progreso. Por favor, espere

1
Laboratorio de BioSistemas
del Departamento de Ingeniería Genética del CINVESTAV Unidad Irapuato Marcadores moleculares: más allá de la reconstrucción filogenética AAAAAAAAAAAAAAA CCCCCCCCCCCCCCCCCC GGGGGGGGGGGGGGGG TTTTTTTTTTTTTTTT Dra. Amanda Castillo Cobián Depto. de Ingeniería Genética CINVESTAV-Irapuato IPN

2
Objetivos 1) Dar a conocer los fundamentos teórico-prácticos de las técnicas más comúnmente utilizadas en el estudio de evolución molecular. Desde la selección de un buen marcador hasta la fechación molecular, pasando por la reconstrucción filogenética como fundamento de todos los estudios en biología evolutiva moderna. 2) Demostrar el tipo de preguntas que pueden resolverse mediante los estudios de evolución molecular y el alcance que pueden tener. 3) Que los participantes adquieran un panorama general sobre los análisis estadísticos y el software disponible para el análisis de datos moleculares. 4) Demostrar el impacto que los estudios evolutivos pueden tener en trabajos aplicados y como han creado un puente de análisis para entender los procesos macro y microevolutivos.
 Demostrar el tipo de preguntas que pueden resolverse mediante los. estudios de evolución molecular y el alcance que pueden tener. 3) Que los participantes adquieran un panorama general sobre los análisis. estadísticos y el software disponible para el análisis de datos moleculares. 4) Demostrar el impacto que los estudios evolutivos pueden tener en. trabajos aplicados y como han creado un puente de análisis para entender. los procesos macro y microevolutivos.")
3
Procesos estocásticos
“ DNA: that registry of chance, that tone-deaf conservatory where the noise is preserved with the music” -Jaques Monod Procesos estocásticos Procesos demográficos El problema del análisis de datos moleculares es distinguir el ruido de lo informativo, lo que guarda algún tipo de información, esto va a depender de las preguntas que queramos contestar y el plazo evolutivo. Historia

4
Análisis de datos moleculares * Proteínas *DNA
Puede cubrir varios aspectos como : Estudios a nivel genética de poblaciones (estructura de poblaciones naturales) Estudios de selección natural (Adaptación molecular) Cálculos de tiempos de divergencia (Relojes moleculares) Todo esto en su conjunto nos da una descripción de los procesos evolutivos que moldearon la historia de los organismos.
 Estudios de selección natural. (Adaptación molecular) Cálculos de tiempos de divergencia. (Relojes moleculares) Todo esto en su conjunto nos da una descripción de los procesos evolutivos que moldearon la historia de los organismos.")
5
A partir de la teoría Darwin-Wallace:
Se generaron dos campos de estudio: la historia evolutiva de los organismos elucidación de las fuerzas evolutivas que moldean la biodiversidad y sus adaptaciones Wallace Darwin

6
La representación de relaciones evolutivas…… es ancestral…...

7
Evolución = Cambio "Evolution is the idea that all existing animals and plants are descended from some one ancestor many millions of years ago or at least a small number of ancestors many millions of years ago” John Maynard Smith

8
Darwin defined evolution as "descent with modification" and
the word 'descent' refers to the way evolutionary modification takes place in a series of populations that are descended from one another.

9
Fuerzas Evolutivas Selección Natural Mutación Deriva Génica Migración
Endogamia

10
¿ Porqué es importante la reconstrucción de las relaciones
Tasa ¿ Porqué es importante la reconstrucción de las relaciones de ancestría-descendencia/ reconstruir la filogenia de los organismos?

11
FILOGENIA Hipótesis estadística de relaciones evolutivas
Se supone que debe de reflejar la historia completa, en la mayoría de los casos esperamos que las genealogías de genes reflejen la historia d ela especie. NO existe la filogenia verdadera, es una hipótesis siempre. Nos habla no sólo de las relaciones sino del proceso…..

12
Reconstrucción filogenética
Metodología que seguimos para estimar relaciones evolutivas… Mirada al pasado…….

13
Relación entre dos especies que han descendido a partir
HOMOLOGÍA: Relación entre dos especies que han descendido a partir de un origen en común, comparten un mismo ancestro, divergencia evolutiva. ANALOGÍA: Relación entre características comúnes, pero no a partir de un ancestro común, no de origen, relación convergente. Datos curiosos: La homología no es una cantidad, se es o no homólogo, no hay tal cosa como un porcentaje de homología. 2) Se dice que dos secuencias poseen un 95% de SIMILITUD!!!!! o identidad. 3) Sólo podemos hacer reconstrucción filogenética con caracteres homólogos!!! Definiciones de homología y analogía. Se es o no homólogo, dejarlo bien claro. Es más adecuado decir identidad o similitud.
 Se dice que dos secuencias poseen un 95% de SIMILITUD!!!!! o identidad. 3) Sólo podemos hacer reconstrucción filogenética con caracteres. homólogos!!! Definiciones de homología y analogía. Se es o no homólogo, dejarlo bien claro. Es más adecuado decir identidad o similitud.")
15
Las relaciones entre los genes homólogos pueden ser del tipo:
Ortólogos – relacionados a un evento de especiación. Parálogos – relacionados a un evento de duplicación génica. Xenólogos – producto de un evento de transferencia horizontal. Definir ortología, paralogía y xenología.

16
Representaciones gráficas de las relaciones evolutivas.

17
Se pueden obtener distintos arboles filogenéticos donde las relaciones
En general, cuando pensamos en la integración del registro fósil, la mayoría de las filogenias son parafiléticas. Se pueden obtener distintos arboles filogenéticos donde las relaciones descritas entre las especies sean: monofiléticas, polifiléticas o parafiléticas.

18
Monofilético Polifilético Ejemplo de relaciones

19
Un árbol filogenético es un diagrama compuesto por nodos y ramas.
nodo terminal nodo interno A B C D E (OTU) Enraizado Rama No todos los arbolitos reflejan tiempo evolutivo, sólo los que son conformados por tasa y tiempo, los de upgma, por ejemplo no necesariamente una rama refleja tiempo evolutivo. Por lo tanto un árbol filogenético es una hipótesis de relaciones evolutivas. Posee dos componentes: tasa y tiempo (la que se acerca más a la descripción real de tiempo evolutivo)
 Enraizado. Rama. No todos los arbolitos reflejan tiempo evolutivo, sólo los que son conformados por tasa y tiempo, los de upgma, por ejemplo no necesariamente una rama refleja tiempo evolutivo. Por lo tanto un árbol filogenético es una hipótesis de relaciones evolutivas. Posee dos componentes: tasa y tiempo (la que se acerca más a la. descripción real de tiempo evolutivo)")
20
Árbol de especies Árboles de especies: contiene un representativo
de cada especie, los nodos se refieren a eventos de especiación. La reconstrucción filogenética es la base de todos estos estudios, es una herramienta que nos permite abordar otro tipo de preguntas, no es un objetivo per se, sino una aproximación a los procesos evolutivos, es dinámica, de ahí la importancia de que se aproxime lo más posible a la realidad.

21
Árboles de genes (genealogías): representan la
historia de un gen en particular, los nodos pueden ser relacionados con eventos de especiación o de duplicación génica. Árbol de genes

22
* Los árboles se construyen en base a las similitudes
entre los distintos OTUs (Unidades taxonómicas). * Estas diferencias o similitudes son cuantificadas con diferentes métodos para generar la topología del árbol basado en las distancias existentes en los organismos. * En las filogenias moleculares la base de la comparación son sustituciones en nucleótidos cuando se trata de DNA o amino ácidos para las proteínas.
. * Estas diferencias o similitudes son cuantificadas con. diferentes métodos para generar la topología del árbol. basado en las distancias existentes en los organismos. * En las filogenias moleculares la base de la comparación. son sustituciones en nucleótidos cuando se trata de DNA. o amino ácidos para las proteínas.")
23
¿Qué es un marcador molecular?
Ser o no ser un marcador molecular……. Ese es el dilema……. O mejor dicho….. ¿Qué es un marcador molecular? Definición de marcador molecular

24
Un marcador molecular es un indicador de la historia evolutiva,
es lo que vamos a analizar para poder dilucidar historia, así como, diferentes procesos evolutivos. Posee información genética y/o fenotípica y sobre todo estadística acerca del proceso evolutivo. Marcadores moleculares utilizados para análisis filogenético: RFLP´S. Fingerprints genómicos (AFLP´s, RAPDS, SNP´s,). Análisis de enzimas multilocus. Secuencias DNA y proteínas.
. Análisis de enzimas multilocus. Secuencias DNA y proteínas.")
25
Características ideales de un marcador molecular:
Suficiente información (estadísticamente hablando) +/- de 500 pb 2) Que posea resolución (dependerá del nivel taxonómico) 3) Que nos relate la historia de la especie (ortólogos, dependiendo del tipo de estudio).
 +/- de 500 pb. 2) Que posea resolución (dependerá del nivel taxonómico) 3) Que nos relate la historia de la especie (ortólogos, dependiendo del tipo de estudio).")
26
Los marcadores moleculares, al ser marcadores del proceso evolutivo,
poseen restricciones de diversos tipos: Sitios catalíticos (función) Exones Intrones Secuencias reguladoras Estructura Posiciones Pseudogenes Diferencias en tasas de sustitución (cambio) Los marcadores moleculares, al ser marcadores del proceso evolutivo, poseen restricciones funcionales y diferencias en tasas de substitución, además de haber sido afectados por los proceso demográficos.
 Exones. Intrones. Secuencias reguladoras. Estructura. Posiciones. Pseudogenes. Diferencias en tasas de sustitución (cambio) Los marcadores moleculares, al ser marcadores del proceso evolutivo, poseen restricciones funcionales y diferencias en tasas de substitución, además de haber sido afectados por los proceso demográficos.")
27
Un árbol filogenético es un diagrama compuesto por nodos y ramas.
nodo terminal nodo interno A B C D E (OTU) Enraizado Rama No todos los arbolitos reflejan tiempo evolutivo, sólo los que son conformados por tasa y tiempo, los de upgma, por ejemplo no necesariamente una rama refleja tiempo evolutivo. Por lo tanto un árbol filogenético es una hipótesis de relaciones evolutivas. Posee dos componentes: tasa y tiempo (la que se acerca más a la descripción real de tiempo evolutivo)
 Enraizado. Rama. No todos los arbolitos reflejan tiempo evolutivo, sólo los que son conformados por tasa y tiempo, los de upgma, por ejemplo no necesariamente una rama refleja tiempo evolutivo. Por lo tanto un árbol filogenético es una hipótesis de relaciones evolutivas. Posee dos componentes: tasa y tiempo (la que se acerca más a la. descripción real de tiempo evolutivo)")
28
Topología con longitudes de ramas (aditiva)
Clados resueltos politomía

29
La reconstrucción filogenética requiere de la estimación
de las relaciones evolutivas, y dependiendo del método de reconstrucción se tendrá también la estimación de la diferencia o divergencia genética. Se estima: Topología Distancia genética Mediante el uso de algoritmos o métodos de optimización. (NJ/UPGMA) (Parsimonia/Verosimilitud) Reconstrucción filogenética requiere de la estimación de las relaciones evolutivas y dependiendo del método de reconstrucción se ten drá también la estimación de la divergencia genética (todo como procesos estadístico) 1) Los algoritmos hacen estimación de topología y distancia, al mismo tiempo. Por lo mismo son muy rápidos. 2) Los métodos basados en un criterio de optimización, hacen una separación y escogen en un espacio de óptimos los mejores, para posteriormente describir el proceso evolutivo como un espacio de probabilidades. Por lo mismo son computacionalmente intensivos y lentos.
 (Parsimonia/Verosimilitud) Reconstrucción filogenética requiere de la estimación de las relaciones evolutivas y dependiendo del método de reconstrucción se ten drá también la estimación de la divergencia genética (todo como procesos estadístico) 1) Los algoritmos hacen estimación de topología y distancia, al mismo tiempo. Por lo mismo son muy rápidos. 2) Los métodos basados en un criterio de optimización, hacen. una separación y escogen en un espacio de óptimos los mejores, para posteriormente describir el proceso evolutivo como un espacio. de probabilidades. Por lo mismo son computacionalmente intensivos. y lentos.")
30
Pero, ¿cómo se determina la homología en los caracteres moleculares?
* Alineación de secuencias (aa ó nucleótidos) -Cada sitio es un caracter con origen compartido (homólogo). Equivale a una característica morfológica. Estadísticamente es más robusto. Para mantener la correspondencia en sitios homólogos es necesario la introducción de ‘gaps’. Indeles Gaps e indeles NO se introducen de manera aleatoria. No interrumpen el marco de lectura y respetan estructura (loops) Alineación, sirve para determinar cuáles son los caracteres homólogos. De ahí que la alineaión es la base de todos los estudios de filogenética. Ya sea de aa o nucleótidos o codones.
 -Cada sitio es un caracter con origen compartido (homólogo). Equivale a una característica morfológica. Estadísticamente es más robusto. Para mantener la correspondencia en. sitios homólogos es necesario la. introducción de ‘gaps’. Indeles. Gaps e indeles NO se introducen de. manera aleatoria. No interrumpen el marco de. lectura y respetan estructura (loops) Alineación, sirve para determinar cuáles son los caracteres homólogos. De ahí que la alineaión es la base de todos los estudios de filogenética. Ya sea de aa o nucleótidos o codones.")
31
Existen diversos programas que realizan alineaciones de secuencias:
ClustalW (y sus variaciones) Muscle T-Coffe Estos programas realizan una alineación global, es decir, fuerzan el alineamiento de las secuencias en su longitud total.
 Muscle. T-Coffe. Estos programas realizan una alineación global, es decir, fuerzan el. alineamiento de las secuencias en su longitud total.")
33
Otros algoritmos de alineación: FASTA BLAST
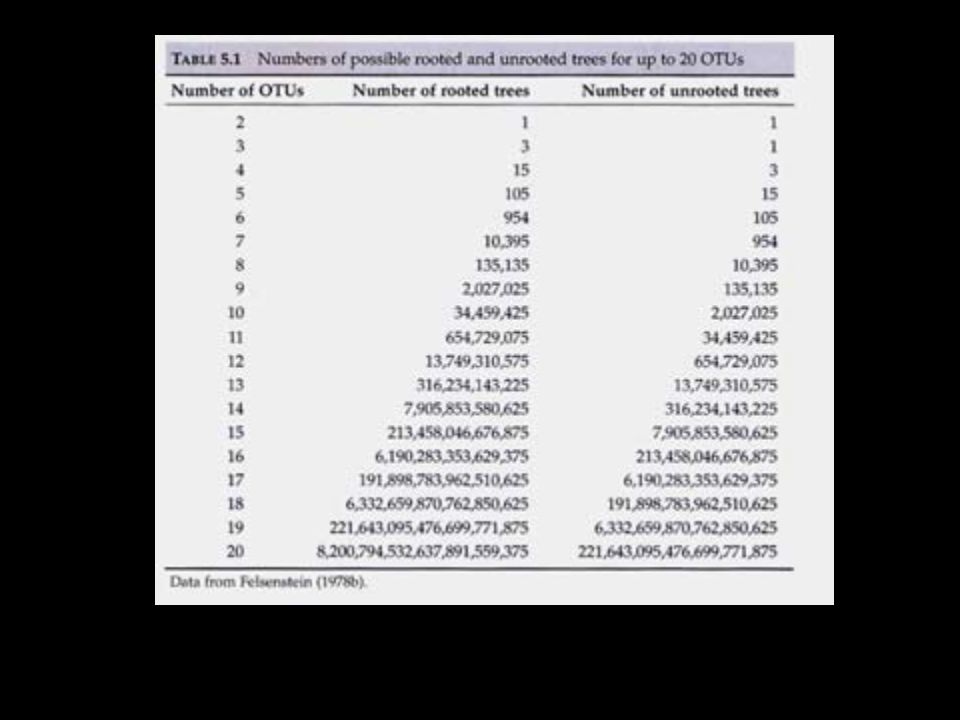
Son programas de alineación local sólo buscan segmentos con la puntuación más alta. (por ejemplo, localizan dominios de proteínas) Poner el número de arbolitos posibles cada vez que se agrega un taxa, y como en el caso de los métodos de distancia esto no es tan importante pero para CO sí lo es.
 Poner el número de arbolitos posibles cada vez que se agrega un taxa, y como en el caso de los métodos de distancia esto no es tan importante pero para CO sí lo es.")
34
A mayor distancia genética es mayor la acumulación de
mutaciones. Dependiendo del tiempo de acumulación de estos cambios puede llegar a ser muy difícil o imposible la alineación en algunas regiones de las secuencias. (Saturación) Las regiones que no son alineables o de homología dudosa deben de ser excluidas de cualquier análisis filogenético, pues meten ruido y generan hipótesis de relaciones falsas.
 Las regiones que no son alineables o de homología dudosa. deben de ser excluidas de cualquier análisis filogenético, pues meten ruido y generan hipótesis de relaciones falsas.")
35
Para los métodos que utilizan un CO, es necesario el desarrollo de
un modelo de sustitución (nucleótidos, aa o codones). Estos modelos son aproximaciones a los procesos naturales de sustitución en el tiempo. Debido a que los métodos con CO ajustan los datos observados a un determinado modelo de sustitución y de acuerdo a esto modelan las diferentes topologías y les brindan un soporte estadístico, es de suma importancia la determinación del modelo de sustitución que más se ajuste a los datos. Los modelos de sustitución describen las probabilidades de cambio ya sea de un nucleótido por otro (A-G, A-C), de un aa por otro Ala-Try, de un codon por otro AGT-GGG, por ejemplo.
. Estos modelos son aproximaciones a los procesos naturales de sustitución. en el tiempo. Debido a que los métodos con CO ajustan los datos observados a un. determinado modelo de sustitución y de acuerdo a esto modelan las. diferentes topologías y les brindan un soporte estadístico, es de suma. importancia la determinación del modelo de sustitución que más se ajuste. a los datos. Los modelos de sustitución describen las probabilidades de cambio. ya sea de un nucleótido por otro (A-G, A-C), de un aa por otro Ala-Try, de un codon por otro AGT-GGG, por ejemplo.")
36
La reconstrucción filogenética requiere de estimaciones de:
Topología Proceso evolutivo (requiere de un modelo de sustitución o cambio) Estos modelos de sustitución nos describen las probabilidades en que se dan las sustituciones. 4) Los métodos que determinan una distancia genética o usan un criterio de optimización ocupan un modelo explícito de sustitución. 5) Los métodos de distancia estiman un parámetro (Número de sust. por sitio) 6) Los métodos con criterio de optimización estiman el valor de cada uno de los parámetros del modelo dada una topología y un grupo de datos. 7) Es necesario usar una prueba estadística para seleccionar el modelo de sustitución que posea un mayor ajuste a nuestros datos.
 Estos modelos de sustitución nos describen las probabilidades en que. se dan las sustituciones. 4) Los métodos que determinan una distancia genética o usan un criterio. de optimización ocupan un modelo explícito de sustitución. 5) Los métodos de distancia estiman un parámetro (Número de sust. por sitio) 6) Los métodos con criterio de optimización estiman el valor de cada uno. de los parámetros del modelo dada una topología y un grupo de datos. 7) Es necesario usar una prueba estadística para seleccionar el modelo de. sustitución que posea un mayor ajuste a nuestros datos.")
37
Modelos de sustitución
A <=> G T <=> C

38
Existen dos aproximaciones para la construcción de modelos
de sustitución: Modelos empíricos = Calculados a partir de la comparación de numerosas alineaciones y que resultan en valores fijos de los parámetros de sustitución. Se utilizan principalmente para AA´s. (Matrices Dayhoff, BLOSUM,etc) 2) Modelos paramétricos = Se basan en el modelado de ciertas propiedades químicas (AA´s) o composicionales, inferidos a partir de cada base de datos. Se utilizan principalmente para nucleótidos y codones (Uso diferencial de codones). Ambos resultan en modelos de procesos Markovianos, definidos por matrices que contienen tasas relativas de ocurrencia de todos los tipos de sustituciones.
 2) Modelos paramétricos = Se basan en el modelado de ciertas. propiedades químicas (AA´s) o composicionales, inferidos a partir. de cada base de datos. Se utilizan principalmente para nucleótidos. y codones (Uso diferencial de codones). Ambos resultan en modelos de procesos Markovianos, definidos por. matrices que contienen tasas relativas de ocurrencia de todos los. tipos de sustituciones.")
39
Ambos métodos describen las tasas relativas, ocurrencia de todos
los tipos de cambio en el tiempo. Se asume que esta matriz es reversible no existe dirección en el tiempo evolutivo, árboles no enraizados, el proceso puede fluir en ambas direcciones. Las sustituciones se describen como resultado de un proceso de mutación al azar, es decir, las mutaciones futuras ocurren al azar y son independientes del estado anterior, dependen únicamente del estado actual (proceso Markoviano). La probabilidad de intercambio de un carácter (mutación) por otro está modelada por una distribución de Poisson.
. La probabilidad de intercambio de un carácter (mutación) por otro. está modelada por una distribución de Poisson.")
40
Se manejan tres tipos de parámetros en el modelado del
proceso de sustitución: Frecuencia (proporción de cada nucleótido en la muestra) Tasas de cambio (transiciones, transversiones, uso de codones) Heterogeneidad en tasas de sustitución (entre posiciones, regiones codificantes VS no codificantes, etc) (Distribución gamma)
 Tasas de cambio (transiciones, transversiones, uso de codones) Heterogeneidad en tasas de sustitución (entre posiciones, regiones codificantes VS no codificantes, etc) (Distribución gamma)")
41
Los diferentes modelos se distinguen por algunos factores básicos:
Frecuencias de nucleótidos = Frecuencia ΠA= ΠG =ΠT= ΠC (JC69, K2P,K3P..) ≠ Frecuencia ΠA ≠ ΠG ≠ ΠT ≠ ΠC (F81, HKY85, Tr93, GTR…) 2) Tasas de transición VS. transversión ti/tv ≠ 0.5 existe un sesgo en algún tipo de sustituciones Generalmente las transiciones son mayores a las transversiones.
 ≠ Frecuencia ΠA ≠ ΠG ≠ ΠT ≠ ΠC (F81, HKY85, Tr93, GTR…) 2) Tasas de transición VS. transversión. ti/tv ≠ 0.5 existe un sesgo en algún tipo de sustituciones. Generalmente las transiciones son mayores a las transversiones.")
42
Los diversos modelos de sustitución se distinguen por su
parametrización: Frecuencia nucleótidos = o ≠ 2) Tasas de sustitución (ti/tv) Tasa 1 Modelo Jukes-Cantor, 1969 Tasa 2 Modelo Kimura-2, F84 Tasa 3 Modelo TrN (2 ti, 1tv) Tasa 6 Modelo GTR (cada sustitución su tasa)
 Tasas de sustitución (ti/tv) Tasa 1 Modelo Jukes-Cantor, Tasa 2 Modelo Kimura-2, F84. Tasa 3 Modelo TrN (2 ti, 1tv) Tasa 6 Modelo GTR (cada sustitución su tasa)")
43
Jukes-Cantor (JC) (Jukes-Cantor, 1969)
Equal base frequencies, all substitutions are equally likely. Equal base frequencies T C A G fN a Rate matrix

44
Jukes-Cantor (un parámetro)
G a a a a C T a

45
Kimura 2-parameters (K80) (Kimura, 1980)
Equal base frequencies, variable transition and transversion frequencies. Equal base frequencies Rate matrix T C A G fN a b

46
Kimura-dos parámetros
G b b b b C T a

47
Felsenstein (F81) (Felsenstein, 1981)
Variable base frequencies, all substitutions equally likely. Variable base frequencies Rate matrix T C A G fT a fC fA fG

48
Hasegawa-Kishino-Yano (HKY85) (Hasegawa,Kishino,Yano, 1985)
Variable base frequencies, variable transition and transversion frequencies. Variable base frequencies Rate matrix T C A G fT a b fC fA fG

49
Tamura-Nei (TN93) (Tamura and Nei, 1993)
Distinguish between two different types of transition (A<=>G) is different to (C <=> T), equal transversion frequencies. Variable base frequencies T C A G fT a b fC fA c fG Rate matrix
 is different to (C <=> T), equal transversion frequencies. Variable base frequencies. T. C. A. G. fT. a. b. fC. fA. c. fG. Rate matrix.")
50
Kimura 3-parameter (K3P)
Variable base frequencies, distinguish between two different types of transvesions (A<=>T) is different to (G <=>C), equal transition frequencies. Variable base frequencies T C A G fN a b c
 is different to (G <=>C), equal transition frequencies. Variable base frequencies. T. C. A. G. fN. a. b. c.")
51
General Time Reversible (GTR) (Tavaré, 1986)
Variable base frequencies, symmetrical substituion matrix. General Time Reversible Variable base frequencies T C A G fT a b c fC d e fA f fG Rate matrix General Time Reversible

52
Modela la heterogeneidad de tasas. Cada sitio posee una tasa tomada
Distribución gamma- para acomodar la heterogeneidad de tasas en la alineación (vs. Sitios invariantes por ej.). Cada sitio tiene una tasa tomadad aleatoriamente e independiente de los demás sitios el parámetro alfa controla la forma de la distribución, alfas arriba de uno es de forma de campana, lo que quiere decir que hay baja heterogeneidad, mientras que debajo de uno es fuerte heterogeneidad. El objetivo de los modelos de suttitución es corregir para múltiples sustituciones. Distribución gamma Modela la heterogeneidad de tasas. Cada sitio posee una tasa tomada aleatoriamente de esta distribución. α controla la forma de la distribución, arriba de 1 es forma de campana, lo que significa baja heterogeneidad en tasas, arriba de uno, parece una L y refleja una gran heterogeneidad de tasas.
. Cada sitio tiene una tasa tomadad aleatoriamente e independiente de los demás sitios el parámetro alfa controla la forma de la distribución, alfas arriba de uno es de forma de campana, lo que quiere decir que hay baja heterogeneidad, mientras que debajo de uno es fuerte heterogeneidad. El objetivo de los modelos de suttitución es corregir para múltiples sustituciones. Distribución gamma. Modela la heterogeneidad de tasas. Cada sitio posee una tasa tomada. aleatoriamente de esta distribución. α controla la forma de la distribución, arriba de 1 es forma de campana, lo que significa baja heterogeneidad en tasas, arriba de uno, parece una L. y refleja una gran heterogeneidad de tasas.")
53
A T G Modelos de substitución de codones
Durante la traducción se involucra el reconocimiento de tripletes o codones que comprenden el Código Genético Universal, de los 64 codones sabemos que tres de ellos son de término, por lo tanto tenemos 61 codones, lo que supone que algunos aminoácidos serán codificados por más de un codón. Debido a la degeneración del código genético, existen tasas de sustitución diferenciales para los codones y por lo consiguiente para cada una de las diferentes posiciones. Existen sustituciones sinónimas (que no cambian el tipo de aa) y no sinónimas que cambian el tipo de aa. 1a 2a 3a A T G dS dN dS dN dN
 y no sinónimas que cambian el tipo de aa. 1a. 2a. 3a. A T G. dS. dN. dS. dN. dN.")
54
UUU Phe UCU Ser UAU Tyr UGU Cys UUC UCC UAC UGC UUA Leu UCA UAA Stop
1* Base Segunda base 3* Base U C A G UUU Phe UCU Ser UAU Tyr UGU Cys UUC UCC UAC UGC UUA Leu UCA UAA Stop UGA UUG UCG UAG UGG Trp CUU CCU Pro CAU His CGU Arg CUC CCC CAC CGC CUA CCA CAA Gln CGA CUG CCG CAG CGG AUU Ile ACU Thr AAU Asn AGU AUC ACC AAC AGC AUA ACA AAA Lys AGA AUG Met ACG AAG AGG GUU Val GCU Ala GAU Asp GGU Gly GUC GCC GAC GGC GUA GCA GAA Glu GGA GUG GCG GAG GGG

55
Ahondar un poco más en los modelos de codones.

56
Modelos de substitución de aminoácidos
PAM matrices- Matriz que describe el cambio en tiempo evolutivo del 1% de los aminoácidos de una proteína. Dayhoff matrices – Se llevo a cabo utilizando 34 superfamilias conocidas de proteínas cercanas entre sí y sus tasas de mutación. BLOSSUM matrices – Utilizan comparaciones entre familias distantes de proteínas y sus valores de similitud.

57
* La reconstrucción filogenética es un proceso de estimación de la
topología y la longitud de las ramas. * Los métodos de reconstrucción filogenética están basados en dos estrategias diferentes: Definición de un algoritmo que determina los pasos a seguir para la reconstrucción de la topología. (Combinan la inferencia de la topología con la mejor topología posible, esto los hace más rápidos) 2) Usando un criterio de optimización que permite decidir cuál de las topologías se ajusta más a nuestros datos. (En este caso topología y su soporte están desacoplados, son más lentos)
 2) Usando un criterio de optimización que permite decidir cuál de. las topologías se ajusta más a nuestros datos. (En este caso topología. y su soporte están desacoplados, son más lentos)")
58
Existen varios métodos de reconstrucción filogenética:
Distancia (Algoritmo) (sustituciones de caracteres) 2) Parsimonia (CO) (estados de carácter, sitios informativos) 3) Máxima verosimilitud (CO) (distribución de probabilidades) 4) Bayesianos (CO)
 (sustituciones de caracteres) 2) Parsimonia (CO) (estados de carácter, sitios informativos) 3) Máxima verosimilitud (CO) (distribución de probabilidades) 4) Bayesianos (CO)")
59
Dentro de estos mismos métodos los más utilizados
históricamente han sido: UPGMA (Parsimonia) 2) NJ (Vecino más cercano) (Distancia) 3) MP (Máxima Parsimonia) (Parsimonia) 4) ML (Máxima verosimilitud) 5)Bayesianos
 2) NJ (Vecino más cercano) (Distancia) 3) MP (Máxima Parsimonia) (Parsimonia) 4) ML (Máxima verosimilitud) 5)Bayesianos.")
60
Los métodos de distancia convierten primero la alineación en
una matriz de distancias. Los más utilizados se basan en el criterio de mínima evolución (UPGMA y NJ). Se apoya el árbol/topología cuya longitud total minimice las distancias entre los otus, a partir de una matriz de distancias pareadas. Poner como se calcula una matriz de distancias con una alineación chiquita.
. Se apoya el árbol/topología cuya longitud total minimice las. distancias entre los otus, a partir de una matriz de distancias. pareadas. Poner como se calcula una matriz de distancias con una alineación chiquita.")
61
Distancias ultramétricas
Usualmente se ajustan a un árbol bajo el supuesto de reloj molecular. Son equidistantes a la raíz del árbol. Las distancias son aditivas. Para un par de secuencias el valor en la matriz corresponde a la suma de longitudes de ramas en el camino más corto que las une (dentro del árbol). En los métodos de evolución mínima se busca el árbol cuya longitud de ramas sea mínima.
. En los métodos de evolución mínima se busca el árbol cuya. longitud de ramas sea mínima.")
62
UPGMA (Unweighted pair-group method with arithmetic mean)
B B B C C D dAB/2 d(AB)C/2 d(ABC)D/2 Tasas de cambio constantes, distancias aritméticas, escala las distancias, reloj molecular, distancias equidistantes, árbol enraizado, topología enraizada se obtiene las longitudes de rama simultáneamente a la topología.
C/2. d(ABC)D/2. Tasas de cambio constantes, distancias aritméticas, escala las distancias, reloj molecular, distancias equidistantes, árbol enraizado, topología enraizada. se obtiene las longitudes de rama simultáneamente a la topología.")
63
Neighbor-joinig method (vecino más cercano)
8 8 7 6 5 4 3 2 1 7 1 2 X 6 X Y 3 5 4 Se escoge el que da la mínima suma de las distancias entre las ramas, minimiza la longitud total del árbol, se obtiene un solo árbol,

64
Máxima Parsimonia (MP)
Busca el árbol que requiere el mínimo número de pasos evolutivos (árbol más parsimonioso) Utiliza el concepto de sitios informativos, todos los demás sitios no son tomados en consideración para la reconstrucción de la topología. Se apoya el árbol con mayor número de sitios informativos. Para el caso de MP la situación se complica pues podemos reconstruir más de un árbol igualmente parsimonioso.
 Utiliza el concepto de sitios informativos, todos los demás sitios no son. tomados en consideración para la reconstrucción de la topología. Se apoya el árbol con mayor número de sitios informativos. Para el caso de MP la situación se complica pues podemos reconstruir más. de un árbol igualmente parsimonioso.")
65
1 A T A T T 2 A T C G T 3 G C A G T 4 G C C G T Este proceso se repite para otros árboles…….

66
1 2 1 2 3 3 4 4

67
Unweighted Parsimony Todas las sustituciones son iguales. Weighted parsimony Hace diferencias entre transiciones y transversiones.

69
Métodos de búsqueda de árboles óptimos:
Exactos .- Garantizan la obtención de un solo árbol. Búsqueda exhaustiva Búsqueda branch-bound 2) Heurísticos (aproximados). – Encontrar soluciones óptimas. Stepwise addition (adición secuencial) Branch swapping (intercambio de ramas) Star decomposition (descomposición a manera de estrella)
 Heurísticos (aproximados). – Encontrar soluciones óptimas. Stepwise addition (adición secuencial) Branch swapping (intercambio de ramas) Star decomposition (descomposición a manera de estrella)")
70
Atracción de ramas largas
Se refiere a situaciones en las que linajes con una tasas de cambio muy diferente al promedio, muestran relaciones con otros linajes a pesar de no ser cierto. Una estrategia para reducir el efecto es agregar más outgroups a nuestra base de datos o secuencias que pudiesen romper estas relaciones aparentes. Explicar el fenómeno y cómo se puede solucionar al agregar más outgroups, por ejemplo.

71
Métodos de Máxima verosimilitud (ML)
Requiere un modelo probabilístico de substitución de nucleótidos, es decir, necesitamos especificar la probabilidad de transición o cambio de un tipo de nucleótido a otro en un intervalo de tiempo para cada rama.

72
Probabilidad de los datos dada una hipótesis
P ( D | H) Hipótesis (H): - árbol (topología) - longitud de ramas - parámetros del modelo de substitución Datos (D): - secuencias de nucleótidos - secuencias de aminoácidos * Método paramétrico que utiliza explícitamente un modelo de sustitución. * Utiliza la matriz original de los datos. * CO para escoger entre árboles
 Hipótesis (H): - árbol (topología) - longitud de ramas. - parámetros del modelo de substitución. Datos (D): - secuencias de nucleótidos. - secuencias de aminoácidos. * Método paramétrico que utiliza explícitamente un modelo de sustitución. * Utiliza la matriz original de los datos. * CO para escoger entre árboles.")
73
i j k l 1 2 3 4 X- nucleótido del nodo ancestral t3 Z t2 Y La probabilidad de encontrar el nucleótido l en la Secuencia 4: Pxl (t1+t2+t3) (tiempo total de X a l, entre los dos nodos) La probabilidad de encontrar el nucleótido Y en el nodo Ancestral de las secs 1, 2, 3 es Pxy (t1) Y así sucesivamente…… t1 X
 (tiempo total de X a l, entre los dos nodos) La probabilidad de encontrar el nucleótido Y en el nodo. Ancestral de las secs 1, 2, 3 es. Pxy (t1) Y así sucesivamente…… t1. X.")
74
Por lo tanto, la probabildiad de tener i, j, k, l en las puntas del árbol se calcula:
Pxl (t1+t2+t3) Pxy (t1) Pyk (t2+t3) Pyz 8t2) Pzi (t3) Pzj (t3) Sólo podemos asumir probabilidades porque en la práctica no conocemos el nucleótido ancestral real. Esta probabilidad se puede inferir a partir de la frecuencia del nucleótido en la muestra real de las secuencias. El cálculo de la función de verosimilitud para una secuencia se puede definir como: n L = P pni Prob (ni mi, t) i=1 La verosimilitud para una secuencia de DNA con un número n de bases que posea la base mi en el sitio i y base ni en el mismo sitio de otra secuencia.
 Pxy (t1) Pyk (t2+t3) Pyz 8t2) Pzi (t3) Pzj (t3) Sólo podemos asumir probabilidades porque en la práctica no conocemos el. nucleótido ancestral real. Esta probabilidad se puede inferir a partir de la frecuencia. del nucleótido en la muestra real de las secuencias. El cálculo de la función de verosimilitud para una secuencia se puede definir como: n. L = P pni Prob (ni mi, t) i=1. La verosimilitud para una secuencia de DNA. con un número n de bases que posea la base mi. en el sitio i y base ni en el mismo sitio de otra. secuencia.")
79
Validación estadística de las reconstrucciones filogenéticas
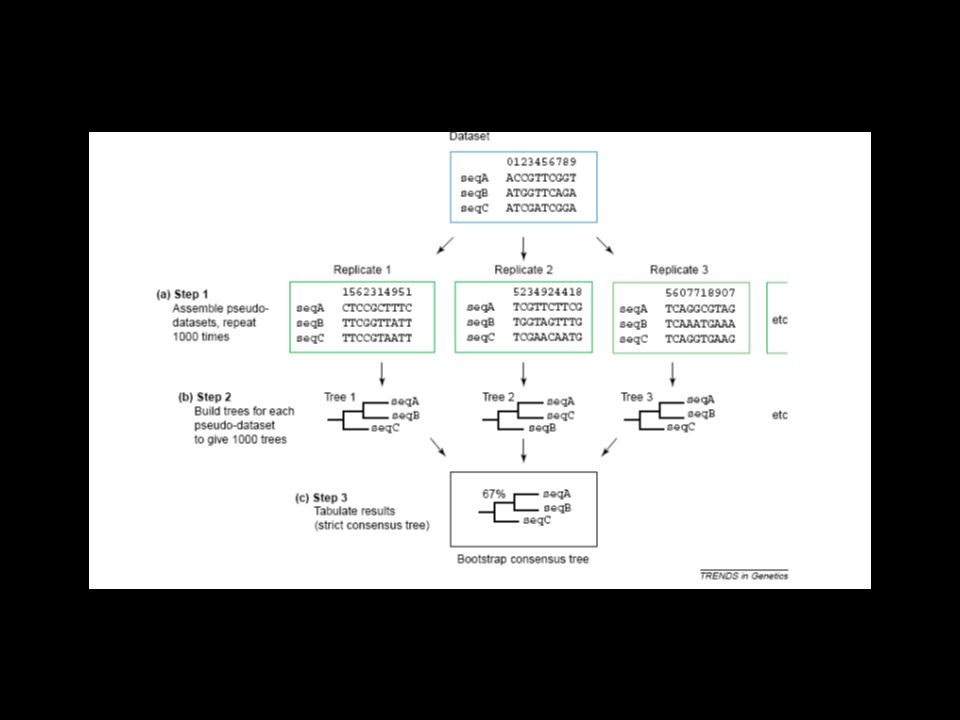
Existen numerosos métodos los más utilizados en la actualidad son: Bootstrap

80
BOOTSTRAP Método estadístico más utilizado.
Se basa en el remuestreo de las secuencias, es decir, aleatoriamente cambia las posiciones de las bases y rehace la construcción filogenética. La medida de soporte estadístico se refiere a un intervalo de confianza de 0 a 100. Supongamos que tenemos la sig. Secuencias: AGTCGGTAA AGTGGGTAA ATCTTGTAA Si cambiamos la posición 7 TAGTCGGAA TAGTGGGAA TATCTTGAA (esta es una submuestra de la distribución original)
")
82
Bootstrapping - an example
Ciliate SSUrDNA - parsimony bootstrap Ochromonas (1) Symbiodinium (2) 100 Prorocentrum (3) Euplotes (8) 84 Tetrahymena (9) 96 Loxodes (4) 100 Tracheloraphis (5) 100 Spirostomum (6) 100 Gruberia (7)
 Symbiodinium (2) 100. Prorocentrum (3) Euplotes (8) 84. Tetrahymena (9) 96. Loxodes (4) 100. Tracheloraphis (5) 100. Spirostomum (6) 100. Gruberia (7)")
83
Inferencia Bayesiana

87
Teorema de Bayes Población hipotética 1 2 60% 40% Población total
Tipo de enfermedad % % Muestreo al azar de 100 individuos (incluyendo grupo I y II) ¿Cuál es la probabilidad que 3 de ellos estén enfermos?
 ¿Cuál es la probabilidad que 3 de ellos estén enfermos")
88
D (probabilidad) definida por:
P= P(D) = P(G1) * P(DI G1) +P(G2) * P(D IG2) 0.6* *0.001= Probabilidad de que un individuo esté enfermo = La probabilidad de no estar enfermo.
 = P(G1) * P(DI G1) +P(G2) * P(D IG2) 0.6* *0.001= Probabilidad de que un individuo esté enfermo = La probabilidad de no estar enfermo.")
89
Probabilidada de que 3 de cada 100 estén enfermos:
P = 100! P3 (1-P)97 / 3!* 97! = BAYES Lo realizamos de manera inversa ¿Cuál es la probabilidad de que un individuo de la muestra al azar que se encuentre enfermo pertenezca al grupo I?
97 / 3!* 97! = BAYES. Lo realizamos de manera inversa. ¿Cuál es la probabilidad de que un individuo de la muestra al azar. que se encuentre enfermo pertenezca al grupo I")
90
PI G1I D I= P(G1)* P(DI G1)/P(D)
0.6*0.01/0.0064=0.94 (Esta es la probabilidad de que pertenezca al grupo I) D - datos – topología de un árbol G1 – clase de sitio, probabilidad de transcición, etc)
 D - datos – topología de un árbol. G1 – clase de sitio, probabilidad de transcición, etc)")
92
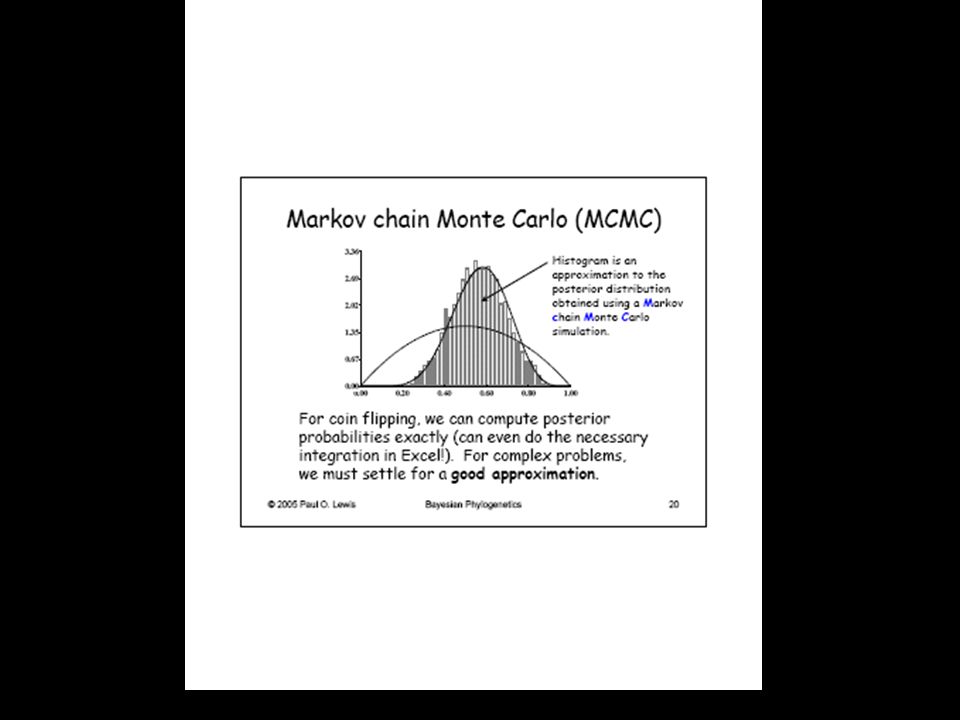
- cadenas markovianas de Monte Carlo
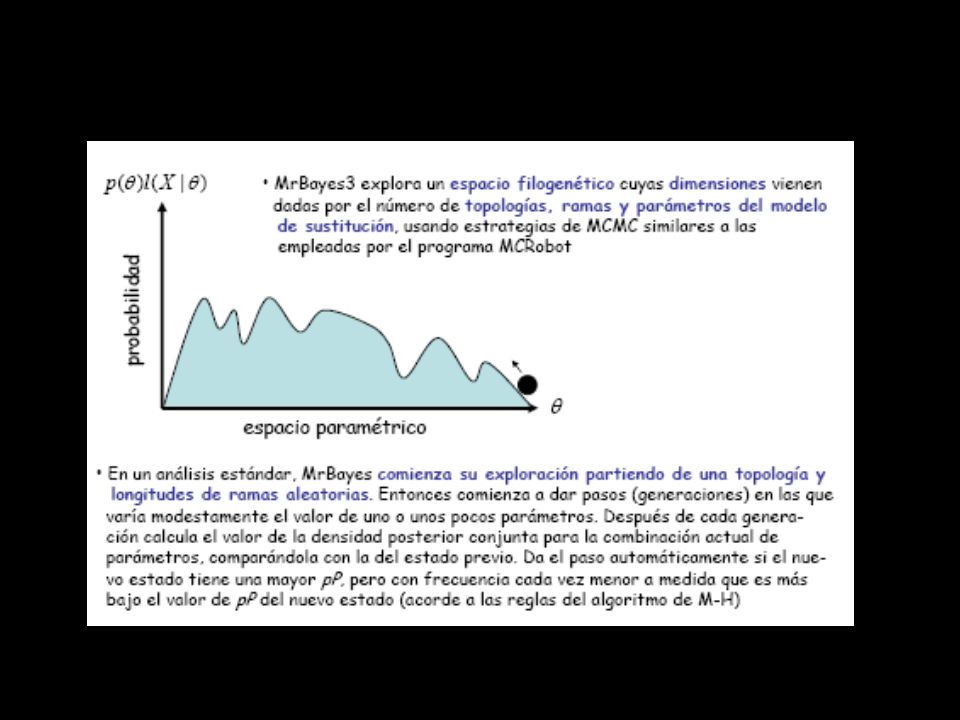
- la probabilidad posterior de un árbol puede interpretarse como la probabilidad de que dicho árbol o clado sea correcto - es imposible estimar dicha pP analíticamente ni siquiera para el caso más simple de 4 OTUs ( (2s - 3)!/2s - 2 (s - 2)! topologías y 2n-3 long. de rama, para arb. no enraiz.) - existen métodos numéricos que permiten aproximar la probabilidad posterior de un árbol (o de cualquier otra hipótesis compleja). El más útil es el de las cadenas markovianas de Monte Carlo (MCMC), implementado en algoritmos como el de Metropolis-Hastings - MCMC se basa en el muestreo de una distribución simulada en vez de calcular dicha distribución mediante integración. Así es posible aproximar el área bajo la curva que representa la distribución de densidad probabilística posterior para inferencias complejas
!/2s - 2 (s - 2)! topologías y 2n-3 long. de rama, para arb. no enraiz.) - existen métodos numéricos que permiten aproximar la probabilidad posterior de un árbol (o de cualquier otra hipótesis compleja). El más útil es el de las cadenas markovianas de Monte Carlo (MCMC), implementado en algoritmos como el de Metropolis-Hastings. - MCMC se basa en el muestreo de una distribución simulada en vez de calcular dicha distribución mediante integración. Así es posible aproximar el área bajo la curva que representa la distribución de densidad probabilística posterior para inferencias complejas.")
99
Impacto de la reconstrucción filogenética:
Cálculo de tiempos de divergencia Selección natural Evolución de la patogénesis Determinación de tasas de sustitución molecular Criminología

Presentaciones similares























 Integrantes: Rubén Levineri Miguel Rozas Juan Yañez Faltan autores y bibliografía.>")








