Descargar la presentación
La descarga está en progreso. Por favor, espere

1
Introducció a la Bioinformàtica
Bioinformàtica: la recerca biomèdica in silico

2
Alineamiento de secuencias

3
Alineamiento de secuencias
Comparar dos (alineación a pares) o más (alineación múltiple) secuencias para buscar una serie de caracteres o patrón de caracteres que están en el mismo orden en las secuencias Secuencia 1: ATGCGACTGACG Secuencia 2: ATGCGACTGACG |||||||||||| Significado de un alineamiento Estadístico Por azar (alineamiento de secuencias al azar pueden producir un 20% identidad) Biológico Comparten un ancestro común. Implica una información funcional, estructural y evolutiva?
 o más (alineación múltiple) secuencias para buscar una serie de caracteres o patrón de caracteres que están en el mismo orden en las secuencias. Secuencia 1: ATGCGACTGACG. Secuencia 2: ATGCGACTGACG. |||||||||||| Significado de un alineamiento. Estadístico. Por azar (alineamiento de secuencias al azar pueden producir un 20% identidad) Biológico. Comparten un ancestro común. Implica una información funcional, estructural y evolutiva")
4
Un alineamiento compara permitiendo:
Alineamiento de secuencias Un alineamiento compara permitiendo: Identificar genes homólogos/similares Asignar funciones biológicas (reales, posibles?) Predecir estructura Encontrar patrones Reconstruir relaciones evolutivas ……
 Predecir estructura. Encontrar patrones. Reconstruir relaciones evolutivas. ……")
5
Homología vs similitud
Homólogo, similar, idéntico Homología dos secuencias son homólogas sólo si derivan de una ancestro común implica una herencia compartida cualitativo se es homólogo o no se es Similitud medida cuantitativa se puede usar una medida de similitud para inferir homología

6
Global vs Local Global: Intentamos alinear todos los caracteres de las secuencias optimizando el número máximo de identidades Local: Alineamos segmentos de las secuencias donde la densidad de identidades es mayor, generamos subalinemientos

7
Alineamiento de secuencias
Un proceso de alineamiento debe efectuar una búsqueda activa del mejor alineamiento posible y debe considerar los cambios que sufren las secuencias: Identidades -> Emparejamientos (match) Sustituciones -> Desemparejamientos (mismatch) Deleciones e inserciones (indel) -> Huecos (gaps)
 Sustituciones -> Desemparejamientos (mismatch) Deleciones e inserciones (indel) -> Huecos (gaps)")
8
Alineamiento de secuencias
Cómo decidir cuál es el mejor? Respuesta: el más significativo desde el punto de vista biológico Pero: necesitamos una medida objetiva Sistemas de puntuación (scoring) reglas para asignar puntos el más simple: match, mismatch, gap Fernán Agüero
 reglas para asignar puntos. el más simple: match, mismatch, gap. Fernán Agüero.")
9
Valoración (score) de un alineamiento
Valoración de un alineamiento Valoración (score) de un alineamiento Máxima puntuación = Alineamiento óptimo F (puntuación emparejamiento idéntico, puntuación emparejamiento similar, puntuación huecos –gap-) Puntuación =
 de un alineamiento. Máxima puntuación = Alineamiento óptimo. F (puntuación emparejamiento idéntico, puntuación emparejamiento similar, puntuación huecos –gap-) Puntuación =")
10
Valoración de un alineamiento
Como valorar un alineamiento? Ejemplo Identidad = 1 Missmatch = 0 Gap = -1 Score = = 6

11
Matriz para DNA Una matriz no es otra cosa que un sistema de scoring que permite asignar puntuaciones individuales a cada una de las letras del alfabeto en uso Fernán Agüero

12
Matriz para DNA A G C T A +1 –3 –3 -3 G –3 +1 –3 -3 C –3 –3 +1 -3
Score Match: + 1 Mismatch: - 3 CAGGTAGCAAGCTTGCATGTCA || |||||||||||| ||||| raw score = 19-9 = 10 CACGTAGCAAGCTTG-GTGTCA Score Match: + 1 Mismatch:

13
Modelos evolutivos DNA
Matriz para DNA Modelos evolutivos DNA Matriz de substitución para DNA Ejemplo: A,A = 1 A,C = 0 C,T = 0,5 Gap = -1 Transiciones / Transversiones p(transición) > p(transversión)
 > p(transversión)")
14
Matrices de puntuación para proteínas
Matriz para proteínas Matrices de puntuación para proteínas Matriz de identidad Secuencia A Tyr Cys Asp Ala Met Secuencia B Phe Met Glu Gly Met Puntuación total del alineamiento: = 1 Matrices de susbtitución Secuencia A Tyr Cys Asp Ala Secuencia B Phe Met Glu Gly Puntuación total del alineamiento: = 4

15
Modelos evolutivos Proteínas
Matriz para proteínas Modelos evolutivos Proteínas Mutaciones (código genético) Substituciones conservativas Matriz de substitución para proteínas PAM 60, 120, 250 (Dayhoff) Extrapolación desde PAM1 BLOSUM 80, 62, 40 Basadas en BLOCKS de secuencias
 Substituciones conservativas. Matriz de substitución para proteínas. PAM 60, 120, 250 (Dayhoff) Extrapolación desde PAM1. BLOSUM 80, 62, 40. Basadas en BLOCKS de secuencias.")
16
Matrices PAM (Percent Accepted Mutation) Evolutionary model
Creadas partiendo de un grupo de secuencias homólogas con un porcentage de similitud igual o superior al 85 %. Proporcionan los cambios esperados entre proteínas homólogas a lo largo de un determinado periodo evolutivo Generan una matriz 20 x 20 Todas calculadas a partir de PAM1 (identidades aprox 99%) Se pueden extrapolar los cambios esperados en periodos cortos a los esperados en periodos largos simplemente multiplicando la matriz original n veces. La más utilizada: PAM 250 (identidades aprox 20%)
 Se pueden extrapolar los cambios esperados en periodos cortos a los esperados en periodos largos simplemente multiplicando la matriz original n veces. La más utilizada: PAM 250 (identidades aprox 20%)")
17
Matrices BLOSUM (BLOck SUbstitution Matrices)
Creadas utilizando un gran número de secuencias NO homólogas pertenecientes a distintas familias Se evaluan las tasas de sustitución en patrones de residuos altamente conservados (BLOCKS) Generan matrices 20 x 20 Las distintas matrices se generan usando agrupaciones de bloques con mayor o menor grado de similitud La matriz por defecto: BLOSUM62
 Generan matrices 20 x 20. Las distintas matrices se generan usando agrupaciones de bloques con mayor o menor grado de similitud. La matriz por defecto: BLOSUM62.")
18
Matriz PAM 250

19
Blosum 62 Negative for less likely substitutions
D C Q E G H I L K M F P S T W Y V X A R N D C Q E G H I L K M F P S T W Y V X F Negative for less likely substitutions D Y F Positive for more likely substitutions Los scores provienen del la observación de los tipos y frecuencias de sustitución en distintas familias proteicas

20
Equivalencia PAM - BLOSUM
BLOSUM: “mejor” para la detección alineamientos locales BLOSUM 62, detección similitudes débiles BLOSUM 45, detección alineamientos largos y débiles

21
PAM vs BLOSUM PAM asume un modelo de evolución markoviano (todos los cambios independientes) BLOSUM no supone ningún modelo evolutivo explícito PAM considera todas las posiciones amino acídicas BLOSUM considera sólo los cambios en posiciones dentro de bloques conservados PAM asume que la distribución de aa de las secuencias que la generaron es representativa de todas las familias de proteínas BLOSUM se generó usando un mayor número de secuencias y de familias distintas PAM requiere un conocimiento previo de la distancia evolutiva Conclusión: PAM es más adecuada para trazar el origen evolutivo de proteínas BLOSUM es más apropiada para hallar dominios conservados

22
Valoración de un alineamiento: gaps
Gap penalty W = g - r(x-1) W es la puntuación x la longitud del gap g la penalización de abrir un gap r la penalizaciónde extender un gap Muchos programas de alineamiento sugieren los valores por defecto Penalización al final del alineamiento Sí en secuencias homólogas misma longitud No en secuencias desconocidas o longitud diferente
 W es la puntuación. x la longitud del gap. g la penalización de abrir un gap. r la penalizaciónde extender un gap. Muchos programas de alineamiento sugieren los valores por defecto. Penalización al final del alineamiento. Sí en secuencias homólogas misma longitud. No en secuencias desconocidas o longitud diferente.")
23
Valoración de un alineamiento: gaps
Valores de penalización de gaps Programas de alineamiento de DNA Programa Tipo de Match Mismatch Penalización Penalización alineamiento score score apertura gaps extensión gaps FASTA local Programas de alineamiento de Proteínas Programa Tipo de Match y Mismatch Penalización Penalización alineamiento score apertura gaps extensión gaps FASTA local Valores BLOSUM BLAST local Valores BLOSUM

24
Valoración de un alineamiento: gaps
Efecto de la variación en la penalización de huecos (gap) (Vingron & Waterman 1994) Si se penaliza alto (relativamente) no aparecerán en el alineamiento Si bajo, gaps serán ubicuos Si la penalización gaps y desemparejamientos (mismatch) es alta habrá un alineamiento local con emparejamientos exactos El alineamiento de proteínas relacionadas muestran que gaps > 5 nunca ocurren
 (Vingron & Waterman 1994) Si se penaliza alto (relativamente) no aparecerán en el alineamiento. Si bajo, gaps serán ubicuos. Si la penalización gaps y desemparejamientos (mismatch) es alta habrá un alineamiento local con emparejamientos exactos. El alineamiento de proteínas relacionadas muestran que gaps > 5 nunca ocurren.")
25
Métodos de alineamiento
Métodos de alineamiento de dos secuencias Dot Matrix (Matriz de puntos) Rápida identificación de indels / No alineamientos óptimos Dynamic Programming (Programación dinámica) Garantiza alineamiento óptimo / Computacionalmente costoso Heuristic Searches (Búsquedas heurísticas) Búsquedas rápidas en bases de datos grandes / alineamientos locales y no siempre óptimos
 Rápida identificación de indels / No alineamientos óptimos. Dynamic Programming (Programación dinámica) Garantiza alineamiento óptimo / Computacionalmente costoso. Heuristic Searches (Búsquedas heurísticas) Búsquedas rápidas en bases de datos grandes / alineamientos locales y no siempre óptimos.")
26
Matriz de puntos (Dot matrix, Gibbs & McIntyre 1970)
Se ponen de manifiesto todos los emparejamientos posibles pero es el investigador quien debe determinar cuales son significativos C| X X G| X T| X : A T T G C T| X | | : | | A|X A T C G C A T C G C A| X C| X - A T G C A G|X X | | | | T| X G A T G C A|- X G A T G C

27
Alineamiento: matriz de puntos
Matriz de puntos (Dot matrix) A|X X X T| X X G| X . T| X . A T C A C T G T A C| X . | | | | | | | A|X X A T C A - - G T A C| X T| X X A|X X A T C A G T A Detección de indels: inserciones / deleciones
 A|X X X T| X X G| X . T| X . A T C A C T G T A C| X . | | | | | | | A|X X A T C A - - G T A C| X T| X X A|X X A T C A G T A. Detección de indels: inserciones / deleciones.")
28
Alineamiento: matriz de puntos
Matriz de puntos de dos proteínas represoras en fagos, λ cI y P22 c2 Nucleótidos Ventana = 11 (longitud del bloque) Astringencia = 7 (Mínima coincidencia admitida) 11/7 ó 15/11 Aminoácidos Ventana = 1 Astringencia = 1 1/1 excepto búsquedas pequeños dominios 15/5
 Astringencia = 7 (Mínima coincidencia admitida) 11/7 ó 15/11. Aminoácidos. Ventana = 1. Astringencia = 1. 1/1 excepto búsquedas pequeños dominios 15/5.")
29
Alineamiento: matriz de puntos
Matriz de puntos de la proteína receptora humana LDL con ella misma. Ventana = 23 Astringencia = 7 Ventana = 1 Astringencia = 1

30
Alineamiento: matriz de puntos
Dot plots sugieren caminos (paths) a través del espacio de alineamientos posibles. Dominios EGF conservados en la urokinse plasminogen activator (PLAU) y el tissue plasminogen activator (PLAT) 90 137 90 137 72 23 23 Path graphs son representaciones más explícitas de un alineamiento. Cada path es un alineamiento único. 72 PLAU 90 EPKKVKDHCSKHSPCQKGGTCVNMP--SGPH-CLCPQHLTGNHCQKEK---CFE 137 PLAT 23 ELHQVPSNCD----CLNGGTCVSNKYFSNIHWCNCPKKFGGQHCEIDKSKTCYE 72
 a través del espacio de alineamientos posibles. Dominios EGF conservados en la urokinse plasminogen activator (PLAU) y el tissue plasminogen activator (PLAT) Path graphs son representaciones más explícitas de un alineamiento. Cada path es un alineamiento único. 72. PLAU 90 EPKKVKDHCSKHSPCQKGGTCVNMP--SGPH-CLCPQHLTGNHCQKEK---CFE 137 PLAT 23 ELHQVPSNCD----CLNGGTCVSNKYFSNIHWCNCPKKFGGQHCEIDKSKTCYE 72.")
31
Programación dinámica
Algoritmos de programación dinámica Métodos computacionales que comparan cada pareja de caracteres y los posicionan de forma que el número de emparejamientos idénticos o relacionados sea el máximo posible Alineamiento global de Needlman-Wunsh (1970) Alineamiento local de Smith-Waterman (1981)
 Alineamiento local de Smith-Waterman (1981)")
32
Programación dinámica
Evalúa y puntúa todos los posibles emparejamientos para cada caracter y lo posiciona en función de dicha puntuación y de la puntuación total de los caracteres ya alineados, construyendo una “ruta” hacia el alineamiento óptimo o de mayor puntuación Uso de un algoritmo recursivo que añade residuos en una posición sobre el alineamiento mejor hasta esa posición. Una ruta óptima que termina en un nodo debe pasar por uno de los tres nodos previos S(i-1,j-1) + c(i,j) S(i,j) = max S(i-1,j) + c(i,-) S(i,j-1) + c(-,j)
 + c(i,j) S(i,j) = max S(i-1,j) + c(i,-) S(i,j-1) + c(-,j)")
33
Programación dinámica
Score nuevo = alineamiento Score alineamiento + previo Score del nuevo emparejamiento A V D S - C V E S L C V D S - V E S L C = -1 B V D S - C Y V E S L C Y V D S - C V E S L C Y = 8

34
Dynamic programming: ejemplo
Un ejemplo: G A T A C T A G A T T A C C A Construir un alineamiento óptimo entre estas dos secuencias Match: Mismatch: Gap: +1 -1 Utilizando las siguientes reglas de scoring: Fernán Agüero

35
Dynamic programming: ejemplo
Ordenar las dos secuencias en una matriz bidimensional G A T A C T A G A T T Los vértices de cada celda se encuentran entre letras (bases). Needleman & Wunsch (1970) A C C A Fernán Agüero
. Needleman & Wunsch (1970) A. C. C. A. Fernán Agüero.")
36
Dynamic programming: ejemplo
T A C T A El objetivo es encontrar la ruta (path) óptimo G A Desde aquí T T A C Hasta acá C A Fernán Agüero
 óptimo. G. A. Desde aquí. T. T. A. C. Hasta acá. C. A. Fernán Agüero.")
37
Dynamic programming: ejemplo
T A C T A Cada path corresponde a un alineamiento único G A T T A C C A Cuál es el óptimo? Fernán Agüero

38
Dynamic programming: ejemplo
T A C T A G El score para una ruta (path) es la suma incremental de los scores de sus pasos (diagonales o lados). A alineada con A A Match = +1 T T A C C A Fernán Agüero
 es la suma incremental de los scores de sus pasos (diagonales o lados). A alineada con A. A. Match = +1. T. T. A. C. C. A. Fernán Agüero.")
39
Dynamic programming: ejemplo
T A C T A El score para una ruta (path) es la suma incremental de los scores de sus pasos (diagonales o lados). G A A alineada con T T Mismatch = -1 T A C C A Fernán Agüero
 es la suma incremental de los scores de sus pasos (diagonales o lados). G. A. A alineada con T. T. Mismatch = -1. T. A. C. C. A. Fernán Agüero.")
40
Dynamic programming: scores: gaps
T A C T A El score para una ruta (path) es la suma incremental de los scores de sus pasos (diagonales o lados). G T alineada con NADA A Gap = -1 T T T alineada con NADA A C C A Fernán Agüero
 es la suma incremental de los scores de sus pasos (diagonales o lados). G. T alineada con NADA. A. Gap = -1. T. T. T alineada con NADA. A. C. C. A. Fernán Agüero.")
41
Dynamic programming: paso a paso (1)
Extender el path paso por paso G A T A C T A -1 G G – G G – -1 +1 A T T +1 -1 -1 A C C A Fernán Agüero

42
Dynamic programming: paso a paso (2)
Incrementar el path paso a paso G A T A C T A -1 -2 G -1 +1 -2 A T Recordar el mejor subpath que lleva a cada punto en la matriz. T A C C A Fernán Agüero

43
Dynamic programming: paso a paso (3)
Incrementar el path paso a paso G A T A C T A -1 -2 G -1 +1 -2 A +2 T Recordar el mejor subpath que lleva a cada punto en la matriz. T A C C A Fernán Agüero

44
Dynamic programming: paso a paso (4)
Incrementar el path paso a paso G A T A C T A -1 -2 G -1 +1 -2 A -2 +2 T Recordar el mejor subpath que lleva a cada punto en la matriz. T A C C A Fernán Agüero

45
Dynamic programming: paso a paso (5)
Incrementar el path paso a paso G A T A C T A -1 -2 -3 G -1 +1 -2 -1 A -2 +2 +1 T Recordar el mejor subpath que lleva a cada punto en la matriz. -3 -1 +1 +3 T A C C A Fernán Agüero

46
Dynamic programming: paso a paso (6)
Incrementar el path paso a paso G A T A C T A -1 -2 -3 -4 -5 G -1 +1 -1 -2 -3 A -2 +2 +1 -1 T Recordar el mejor subpath que lleva a cada punto en la matriz. -3 -1 +1 +3 +2 +1 T -4 -2 +2 +2 +1 A -5 -3 -1 +1 +3 +2 C C A Fernán Agüero

47
Dynamic programming: paso a paso (7)
Incrementar el path paso a paso G A T A C T A +1 -1 -2 +2 -4 -3 +3 -8 -7 -6 -5 +4 G A T Recordar el mejor subpath que lleva a cada punto en la matriz. T A C C A Fernán Agüero

48
Dynamic programming: best path
Recorrer el camino de atrás hacia adelante para obtener el mejor path y alineamiento. G A T A C T A +1 -1 -2 +2 -4 -3 +3 -8 -7 -6 -5 +4 G A T T A C C A Fernán Agüero

49
Dynamic programming: alineamiento obtenido
Imprimir el alineamiento A A - T C G T T A C C A Fernán Agüero

50
Dynamic programming: Smith-Waterman
El método fue modificado (Smith-Waterman) para obtener alineamientos locales El método garantiza la obtención de un alineamiento óptimo (cuyo score no puede ser mejorado) La complejidad es proporcional al producto de las longitudes de las secuencias a alinear Fernán Agüero
 para obtener alineamientos locales. El método garantiza la obtención de un alineamiento óptimo (cuyo score no puede ser mejorado) La complejidad es proporcional al producto de las longitudes de las secuencias a alinear. Fernán Agüero.")
51
Programación dinámica
Preparación de una matriz n x m Alineamiento global de Needlman-Wunsh S(i-1,j-1) + c(i,j) S(i,j) = max S(i-1,j) + c(i,-) S(i,j-1) + c(-,j)
 + c(i,j) S(i,j) = max S(i-1,j) + c(i,-) S(i,j-1) + c(-,j)")
52
Ejemplo on-line: alineamiento global vs local
Preparación de una matriz n x m

53
M | N G A - L - S | D | R | T | - T - E - T
Ejemplo on-line: alineamiento global vs local M | N G A - L - S | D | R | T | - T - E - T

54
Ejemplo on-line: alineamiento global vs local
D R - T G E |

55
Ejemplo on-line: alineamiento global vs local
D R - T G E |

56
Ejemplo on-line: alineamiento global vs local
D R - T G E |

57
Ejemplo on-line: alineamiento global vs local
D R - T G E |

58
Ejemplo on-line: alineamiento global vs local
D R T - G E |

59
Programación dinámica
Alineamiento 1 sequence 1 M - N A L S D R T sequence 2 M G S D R T T E T score 0 -3 1 0 -1 3 = -5 Alineamiento 2 sequence 1 M N - A L S D R T sequence 2 M G S D R T T E T score 6 0 1 0 -1 3 = -6 Alineamiento 3 (no penalización de gap final) sequence 1 M N A L S D R T - - - sequence 2 - - M G S D R T T E T score 0 2 4 6 3 0 0 0 = 10
 sequence 1 M N A L S D R T sequence M G S D R T T E T score = 10.")
60
Programación dinámica
Alineamiento local con el algoritmo de Smith-Waterman Debe haber puntuación negativa para los desemparejamientos y si la puntuación de la matriz obtiene un valor negativo se pone 0 Así el alineamiento puede empezar en cualquier punto y se acaba cuando la puntuación alcanza el valor de 0 secuencia 1 S D R T secuencia 2 S D R T score = 15

61
Alineamiento global y local
Smith & Waterman (1981) Las secuencias se alinean en regiones pequeñas y aisladas Needleman & Wunsch (1970) Las secuencias se alinean esencialmente de un extremo a otro
 Las secuencias se alinean en regiones pequeñas y aisladas. Needleman & Wunsch (1970) Las secuencias se alinean esencialmente de un extremo a otro.")
62
Búsquedas por similitud
Heuristic searches MPKRSEYRQGTPNWVDLQTTDQSAAKKFYTLFGWGYDDNPVPGGGGVYSMATLNGEAVAAIAPMPPGAPEGMPPIWNTYIAVDDVDAVVDKVVPGGGQVMMPAFDIGDAGRMSFITDPTGAAVGWQANRHIGATLVNETGTLIWNELLTDKPDLALAFYEAVVGLTHSSMEIAAGQNY ¿Hay en la base de datos alguna secuencia similar a mi secuencia problema? Búsquedas por similitud Resultados Similar to ………… Unknown but similar to sevral hypothetical proteins from… Putative hypothetical protein……..

63
Búsqueda de similares en una base de datos
Objetivo: comparar una secuencia frente a una base de datos, comprar doss base de datos,.. Algoritmos Exactos: Smith-Waterman (sssearch, lalign, ..) Heurísticos: BLAST (búsqueda de “words” similares) FASTA (búsqueda de “k-tuplos” idénticos)
 Heurísticos: BLAST (búsqueda de words similares) FASTA (búsqueda de k-tuplos idénticos)")
64
Métodos heurísticos BLAST (Basic Local Alignment Search Tool) FASTA
Método heurístico: prueba y error Suele encontrar secuencias relacionadas pero nunca hay garantía absoluta. Ventaja: 50 veces más rápido que programas dinámicos Usos: búsquedas sobre bases de datos de gran tamaño BLAST (Basic Local Alignment Search Tool) (Altschul, et al, 1990, J Mol Biol, 215:403-10) Concentra la búsqueda en patrones cortos más significativos, (palabra, word size: 3 aa / 11 nt). Rápido, menos sensible? FASTA (Lipman y Pearson, 1985; Pearson y Lipman, 1988) El algoritmo busca patrones cortos consecutivos (palabras o k-tuplos, k= 1-2 aa / 4-6 nt) entre la secuencia problema y las de la base de datos.
 (Altschul, et al, 1990, J Mol Biol, 215:403-10) Concentra la búsqueda en patrones cortos más significativos, (palabra, word size: 3 aa / 11 nt). Rápido, menos sensible FASTA. (Lipman y Pearson, 1985; Pearson y Lipman, 1988) El algoritmo busca patrones cortos consecutivos (palabras o k-tuplos, k= 1-2 aa / 4-6 nt) entre la secuencia problema y las de la base de datos.")
65
Secuencias problema Búsquedas con secuencias de DNA o aa
FASTA, BLAST >SeqDNA_Prob1.seq ATGAAGGACTTAGTCGATACCACAGAGATGTACTTGCGTACTATCTATGAGCTGGAAGAAGAGGGAGTCACCCCTCTTCGCGCTAGGATCGCTGAGCG Búsquedas con perfiles Archivos con alineamientos D-HQSNGA ESHQ-YTM EAHQSN-L EGVQSYSL Búsquedas con Blocks Motivos alineados sin gaps (PSSM position-specific scoring matrix) DAHQSN ESHQSY EAHQSN EGVQSY Búsquedas de patrones y motivos PROSITE, INTERPRO, PFAM, .. DAHQSN
 DAHQSN. ESHQSY. EAHQSN. EGVQSY. Búsquedas de patrones y motivos. PROSITE, INTERPRO, PFAM, .. DAHQSN.")
66
Significación - Valoración
de los alineamientos Valoración mediante matrices Penalización por disimilitud (aparición y extensión de gaps). Este valor es la Puntuación bruta (raw score, Sraw). Puntuación de bits (Sbit). Permite comparar resultados obtenidos por diferentes sistemas. Se introducen parámetros utilizados por el programa de alineamiento. E value alineamiento debido al azar. Los valores más cercanos a cero indican una mayor relevancia. Se puede considerar relevante cuando E < 0.05.
. Este valor es la Puntuación bruta (raw score, Sraw). Puntuación de bits (Sbit). Permite comparar resultados obtenidos por diferentes sistemas. Se introducen parámetros utilizados por el programa de alineamiento. E value alineamiento debido al azar. Los valores más cercanos a cero indican una mayor relevancia. Se puede considerar relevante cuando E <")
67
E = numero de hits esperado por azar
Significancia Expect Value E = numero de hits esperado por azar Un E-value de 10 significa que, en una base de datos de igual tamaño, se pueden encontrar 10 alineamientos con la misma puntuación por simple azar. Valores mas bajos serán mas significativos

68
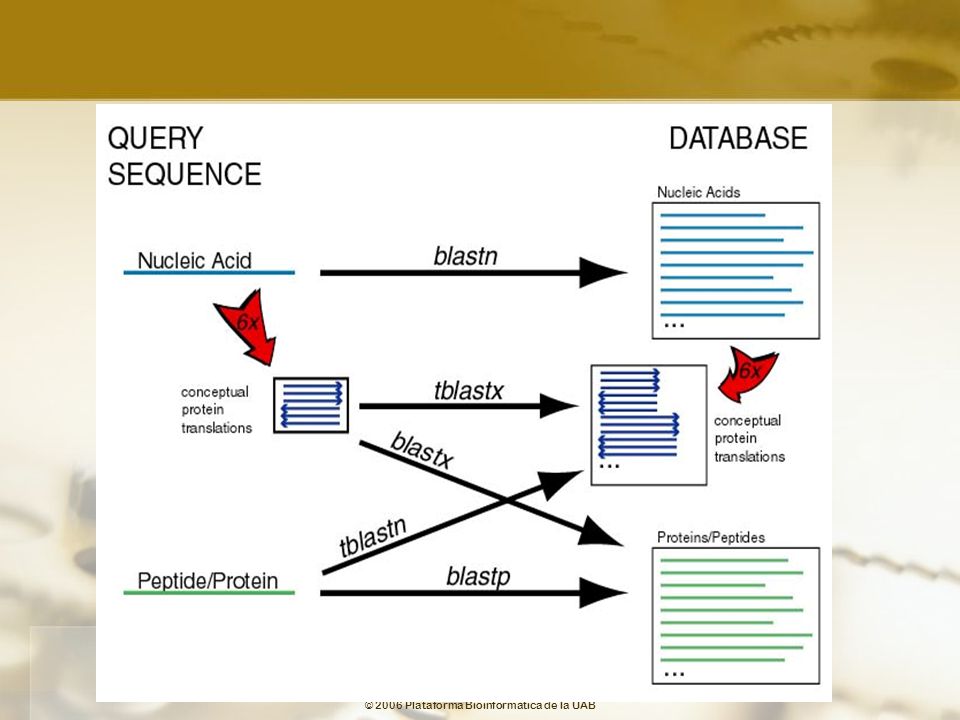
(Basic Local Alignment Search Tool)
Altschul, et al, 1990, J Mol Biol, 215:403-10 Heurístico BLAST intenta encontrar muchos matches sacrificando la especificidad por la velocidad. Se pueden perder apareamientos. Estrategia BLAST Búsqueda de proteínas mediante criterios de semejanza (no identidad). Se utilizan matrices de semejanza Trabaja con segmentos pequeños que permiten acelerar el proceso (High-scoring segment pair) Extensión de los match hacia los lados Rápido y sensible Usos búsquedas en las bases de datos alineamiento de pares de secuencias Glossary
. Se utilizan matrices de semejanza. Trabaja con segmentos pequeños que permiten acelerar el proceso. (High-scoring segment pair) Extensión de los match hacia los lados. Rápido y sensible. Usos. búsquedas en las bases de datos. alineamiento de pares de secuencias. Glossary.")
69
Esquema BLAST

70
Nucleótidos Query: GTACTGGACATGGACCCTACAGGAA GTACTGGACAT
Word Size = 11 GTACTGGACAT TACTGGACATG ACTGGACATGG CTGGACATGGA TGGACATGGAC GGACATGGACC GACATGGACCC ACATGGACCCT Minimum word size = 7 blastn default = 11 megablast default = 28

71
Proteínas GTQITVEDLFYNIATRRKALKN Query: GTQ TQI QIT ITV TVE VED EDL
Word Size = 3 GTQ TQI QIT ITV TVE VED EDL DLF ... Similares LTV, MTV, ISV, LSV, etc.

73
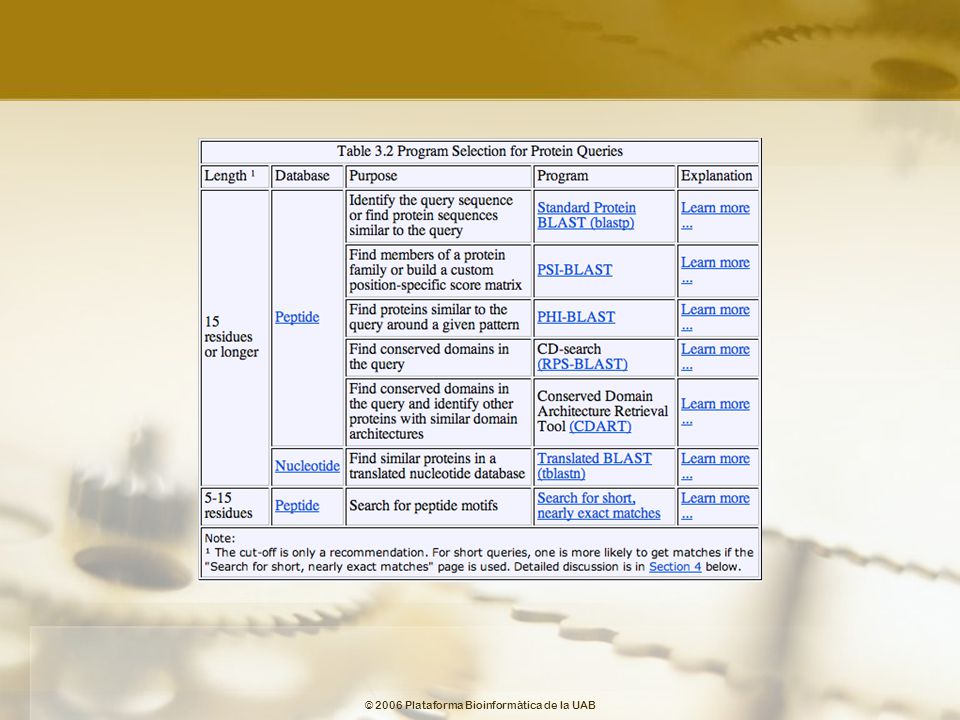
Selección del programa
BLAST Help

77
Conserved Domain Database
Opciones Conserved Domain Database

78
Introducción de la Secuencia
FASTA FORMAT

79
Otras opciones Expect: 10, máximo de 10 al azar. Valores inferiores son mas restrictivos. Word Size: medida de los fragmentos (k-tup FASTA)
 .")
80
Formatos

81
Práctica Objetivo general BLAST DNA
Buscar secuencias similares a las secuencias problema. BLAST DNA Copiar la Secuencia PROBLEMA i someterla a BLAST de DNA >SeqDNA_Prob1.seq ATGAAGGACTTAGTCGATACCACAGAGATGTACTTGCGTACTATCTATGAGCTGGAAGAAGAGGGAGTCA CCCCTCTTCGCGCTAGGATCGCTGAGCGTCTGGAACAATCTGGACCTACAGTTAGCCAAACCGTTGCCCG TATGGAGCGCGATGGACTTGTCGTTGTCGCCTCAGACCGCAGTCTACAAATGACACCGACAGGCCGCACT TTAGCGACTGCAGTTATGCGTAAACATCGCTTAGCTGAGCGCCTTCTTACCGATATCATTGGTCTAGATA TCAATAAAGTTCACGATGAAGCCTGCCGCTGGGAACACGTTATGAGTGACGAAGTTGAACGCAGGCTCGT GAAAGTATTGAAAGATGTCAGTCGGTCCCCCTTCGGAAACCCAATTCCAGGTCTCGACGAACTCGGCGTA GGCAATTCTGACGCGGCAGCCCCCGGAACTCGCGTTATTGACGCTGCCACCAGCATGCCCCGCAAAGTAC GCATTGTTCAGATTAACGAAATCTTTCAAGTTGAAACGGATCAGTTTACACAGCTCCTCGATGCTGACAT CCGTGTTGGATCAGAAGTCGAAATTGTAGATAGAGACGGCCACATCACGTTGAGCCACAATGGAAAAGAT GTCGAACTCCTCGATGATCTGGCTCACACTATTCGTATCGAAGAACTCTAA Iniciar una sesión BLAST Nucleotide Limitar la búsqueda a Blast de Bacteria

82
Práctica BLAST Proteína
Copiar la secuencia PROBLEMA i someterla a BLAST de Proteína >SeqProt-Prob1.pep MENRIDRIKKQLHSSSYKLTPQREATVRVLLENEEDHLSAEDVYLLVKEKSPEIGLATVY RTLELLTELKVVDKINFGDGVSRYDLRKEGAAHFHHHLVCMEFGAVDEIEGDLLEDVEEI IERDWKFKIKDHRLTFHGICHRCNGKETE Iniciar una sesión BLAST Protein Limitar la búsqueda a Blast de Bacteria Ejecutar Blast

83
Proteína x DNA traducido a Proteína
Práctica t n Proteína x DNA traducido a Proteína Objetivo Pretendemos encontrar secuencias bacterianas en diferentes genomas de microorganismos. Realizar una búsqueda tBLASTn sobre genomas microbianos utilizando la secuencia: >Proteína problema MPKRSEYRQGTPNWVDLQTTDQSAAKKFYTSLFGWGYDDNPVPGGGGVYSMATLNGEAVAAIAPMPPGAP EGMPPIWNTYIAVDDVDAVVDKVVPGGGQVMMPAFDIGDAGRMSFITDPTGAAVGLWQANRHIGATLVNE TGTLIWNELLTDKPDLALAFYEAVVGLTHSSMEIAAGQNYRVLKAGDAEVGGCMEPPMPGVPNHWHVYFA VDDADATAAKAAAAGGQVIAEPADIPSVGRFAVLSDPQGAIFSVLKPAPQQ

84
DNA traducido a Proteína x Proteína
Práctica X DNA traducido a Proteína x Proteína Objetivo Detectar similares a la secuencia problema y determinar si hay errores. Realizar una búsqueda con BLASTx usando: >DNA desconocido ATGCCCAAGAGAAGCGAATACAGGCAAGGCACGCCGAACTGGGTCGACCTTCAGACCACCGATCAGTCCG CCGCCAAAAAGTTCTACACATCGTTggtGTTCGGCTGGGGTTACGACGACCCGGTCCCCGGAGGCGGTGG GGTCTATTCCATGGCCACGCTGAACGGCGAAGCCGTGGCCGCCATCGCACCGATGCCCCCGGGTGCACCG GAGGGGATGCCGCCGATCTGGAACACCTATATCGCGGTGGACGACGTCGATGCGGTGGTGGACAAGGTGG TGCCCGGGGGCGGGCAGGTGATGATGCCGGCCTTCGACATCGGCGATGCCGGCCGGATGTCGTTCATCAC CGATCCGACCGGCGCTGCCGTGGGCCTATGGCAGGCCAATCGGCACATCGGAGCGACGTTGGTCAACGAG ACGGGCACGCTCATCTGGAACGAACTGCTCACGGAttgGCCGGATTTGGCGCTAGCGTTCTACGAGGCTG TGGTTGGCCTCACCCACTCGAGCATGGAGATAGCTGCGGGCCAGAACTATCGGGTGCTCAAGGCCGGCGA CGCGGAAGTCGGCGGCTGTATGGAACCGCCGATGCCCGGCGTGCCGAATCATTGGCACGTCTACTTTGCG GTGGATGACGCCGACcccACGGCGGCCAAAGCCGCCGCAGCGGGCGGCCAGGTCATTGCGGAACCGGCTG ACATTCCGTCGGTGGGCCGGTTCGCCGTGTTGTCCGATCCGCAGGGCGCGATCTTCAGTGTGTTGAAGCC CGCACCGCAGCAATAG

85
Estrategia FASTA k-tup DNA: 6 Proteína: 2 Alineamientos locales
FASTA utiliza una matriz de substitución sólo durante la fase de extensión La reducción del tiempo de búsqueda conlleva una pérdida de sensibilidad y selectividad Estrategia Búsqueda de zonas comunes por identidad y sin gaps. Uso de k-tuplo. Velocidad y sensibilidad determinadas por la longitud de la palabra usada. Las palabras cortas hacen la búsqueda más lenta y sensible. Valoración de los match por similitud y unión de las regiones con gaps Evaluación exhaustiva de los mejores alineamientos k-tup DNA: 6 Proteína: 2

86
Programas FASTA Programa Funciones fasta3 tfasta3* ssearch* fastx/y3
DNA, proteína frente bd DNA y bd Prot (fasta) y proteínas frente bd DNA traducido (tfasta), no admite frameshifts DNA, proteína frente bd DNA y bd Prot mediante el algoritmo Smith-Waterman Muy lento <10 fasta pero muy sensible. fastx/y3 DNA (traducido 3 frames) frente bd proteínas. Admite gaps y frameshifts Velocidad x > y tfastx/y3 Proteína frente bd DNA traducido 6 frames fasts3 tfasts3* Mezcla de péptidos pequeños relacionados (mass-spec) frente bd proteína (fasts) o bd DNA (tfasts)* fastf3 tfastf3* Mezcla de péptidos (obtenida por Edman o CNBr) frente bd proteína (fastf) o bd DNA (tfastf)* >mgstm1 MILG, MLLEYTD, MGDAP >mgstm1 MGCEN, MIDYP, MLLAY, MLLGY
 y proteínas frente bd DNA traducido (tfasta), no admite frameshifts. DNA, proteína frente bd DNA y bd Prot mediante el algoritmo Smith-Waterman. Muy lento <10 fasta pero muy sensible. fastx/y3. DNA (traducido 3 frames) frente bd proteínas. Admite gaps y frameshifts. Velocidad x > y. tfastx/y3. Proteína frente bd DNA traducido 6 frames. fasts3. tfasts3* Mezcla de péptidos pequeños relacionados (mass-spec) frente bd proteína (fasts) o bd DNA (tfasts)* fastf3. tfastf3* Mezcla de péptidos (obtenida por Edman o CNBr) frente bd proteína (fastf) o bd DNA (tfastf)* >mgstm1. MILG, MLLEYTD, MGDAP. >mgstm1. MGCEN, MIDYP, MLLAY, MLLGY.")
87
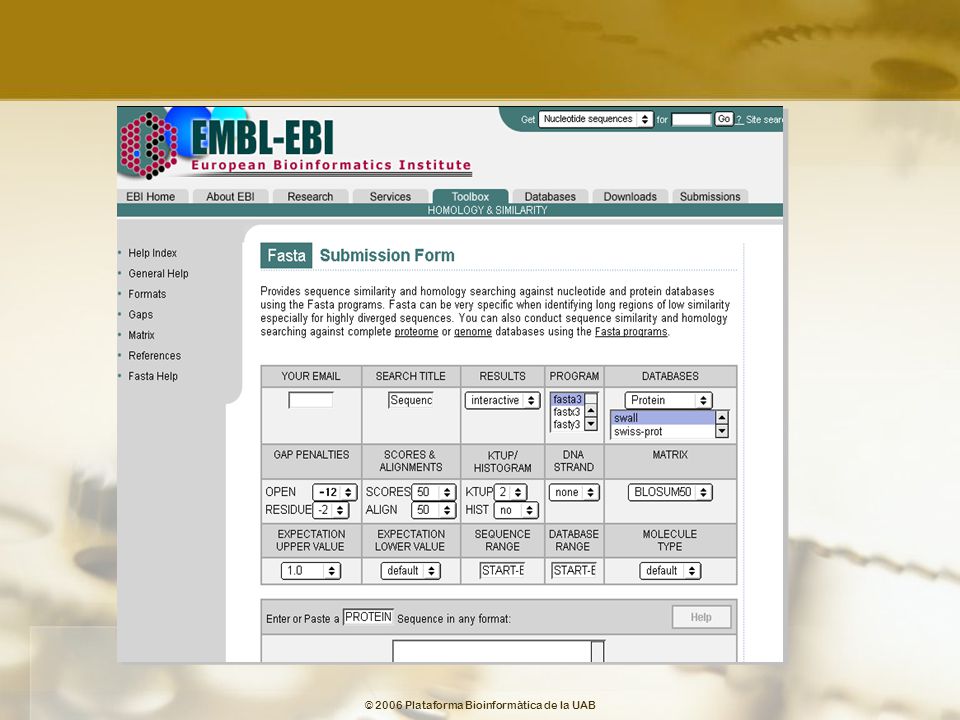
Opciones FASTA MATRIX Matriz de búsqueda GAP PENALTIES
GAPOPEN: Penalización por apertura gap (-12 para proteínas, -16 para DNA). GAPEXT: Penalización extensión del gap (-2 para proteínas, -4 para DNA). HISTOGRAM Muestra el histograma con las frecuencias de las coincidencias por azar. SCORES Puntuación de los alineamientos ALIGNMENTS Número de alineamientos que se visualizaran KTUP Proteínas: 2 DNA: 6 STRAND Cadena de DNA EXPECTATION VALUE Límite superior para la presentación de un alineamiento. Valores por defecto son 10.0 para Prot/Prot; 5.0 para proteínas frente Prot/Prot de DNA y 2.0 para búsquedas DNA/DNA. EXPECTATION VALUE THRESHOLD Límite inferior para la presentación de un alineamiento.
. GAPEXT: Penalización extensión del gap (-2 para proteínas, -4 para DNA). HISTOGRAM. Muestra el histograma con las frecuencias de las coincidencias por azar. SCORES. Puntuación de los alineamientos. ALIGNMENTS. Número de alineamientos que se visualizaran. KTUP. Proteínas: 2 DNA: 6. STRAND. Cadena de DNA. EXPECTATION VALUE. Límite superior para la presentación de un alineamiento. Valores por defecto son 10.0 para Prot/Prot; 5.0 para proteínas frente Prot/Prot de DNA y 2.0 para búsquedas DNA/DNA. EXPECTATION VALUE THRESHOLD. Límite inferior para la presentación de un alineamiento.")
89
Job FASTA

90
Resultados FASTA

91
Mview Results FASTA

92
Visual FASTA

93
Práctica FASTA Búsqueda por similitud Iniciar una sesión FASTA
Seleccionar el programa y fijar los parámetros de búsqueda en función de la naturaleza de la secuencia problema. Introducir la secuencia problema Someter la búsqueda Visualizar y comentar los resultados Diferencias frente a BLAST

95
Comparación múltiple de secuencias Alineamiento múltiple = Tabla 2D
1 2 3 4 5 6 7 8 9 10 I Y D G A V - E L II III F IV Q Cons y d A/I V/L e l Alineamiento múltiple = Tabla 2D Identificación de regiones conservadas Predicción de estructuras y funciones Diseño de experimentos para probar y modificar funciones de proteínas concretas Identificación de nuevos miembros de una familia de proteínas

96
Comparación múltiple de secuencias
FHIT_HUMAN MS-F RFGQHLIKP-SVVFL KTELSFALVNRKPVV PGHVLV... APH1_SCHPO MPKQ LYFSKFPVG-SQVFY RTKLSAAFVNLKPIL PGHVLV... HNT2_YEAST MILSKTKKPKSMNKP IYFSKFLVT-EQVFY KSKYTYALVNLKPIV PGHVLI... Y866_METJA MCIF CKIINGEIP-AKVVY EDEHVLAFLDINPRN KGHTLV... Un método de alineamiento múltiple verdadero, debería alinear todas las secuencias al mismo tiempo. Pero no existe un método computacional que pueda realizar esto en tiempo razonable

97
Cómo se resuelve un alineamiento múltiple
de 3 secuencias? Usando Prgramación dinámica en una matriz tridimensional Objetivo: encontrar el camino óptimo

98
Complejidad del algoritmo de Programación Dinámica (PD)
El número de comparaciones que el PS tiene debe realizar para llenar la matriz (sin usar heurísticas y excluyendo gaps) es el producto de las longitudes de las dos secuencias (N x M) La complejidad del algoritmo crece en forma exponencial con el número de secuencias Alinear dos secuencias de 300 nt implica realizar 300 x 300 = 90,000 comparaciones Alinear tres secuencias de 300 nt implica realizar 300 x 300 x 300 = 27,000,000 comparaciones!!
 es el producto de las longitudes de las dos secuencias (N x M) La complejidad del algoritmo crece en forma exponencial con el número de secuencias. Alinear dos secuencias de 300 nt implica realizar. 300 x 300 = 90,000 comparaciones. Alinear tres secuencias de 300 nt implica realizar. 300 x 300 x 300 = 27,000,000 comparaciones!!")
99
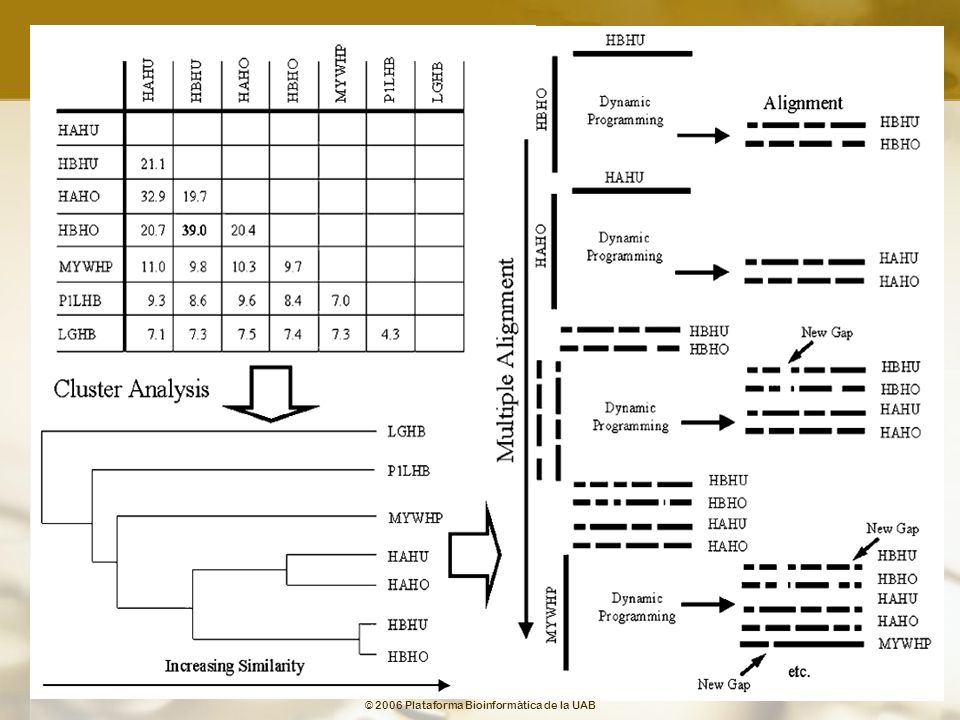
Aproximaciones al algoritmo de Programación Dinámica
Alinear todas las secuencias por pares Usar los scores para construir un árbol Alinear progresivamente (siguiendo el orden que sugiere el árbol) todas las secuencias para producir un Alineamiento Múltiple No es un verdadero Alineamiento Múltiple Las secuencias se alinean por pares
 todas las secuencias para producir un Alineamiento Múltiple. No es un verdadero Alineamiento Múltiple. Las secuencias se alinean por pares.")
101
Programa de alineamiento múltiple Alineamiento progresivo
Clustal W Thompson J.D., Higgins D.G., Gibson T.J. (1994) "CLUSTAL W: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice."; Nucleic Acids Res. 22: Programa de alineamiento múltiple Alineamiento progresivo
 CLUSTAL W: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. ; Nucleic Acids Res. 22: Programa de alineamiento múltiple. Alineamiento progresivo.")
102
Clustal W Estrategia general Alineamiento rápido Alineamiento múltiple
Thompson J.D., Higgins D.G., Gibson T.J. (1994) "CLUSTAL W: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice."; Nucleic Acids Res. 22: Estrategia general Alineamiento rápido obtención de las mejores parejas análisis de clusters creación de un árbol guía Alineamiento múltiple se utiliza el árbol guía anterior optimización alineamiento de los pares más próximos introducción de gaps para mejorar el alineamiento alineamiento de las parejas optimización mediante inclusión de nuevos gaps
 CLUSTAL W: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. ; Nucleic Acids Res. 22: Estrategia general. Alineamiento rápido. obtención de las mejores parejas. análisis de clusters. creación de un árbol guía. Alineamiento múltiple. se utiliza el árbol guía anterior. optimización alineamiento de los pares más próximos. introducción de gaps para mejorar el alineamiento. alineamiento de las parejas. optimización mediante inclusión de nuevos gaps.")
103
Opciones Generales Clustal W
YOUR SEARCH TITLE CPU MODE clustalw_mp - multiprocessor SGI systems. clustalw - genérico (CPU simple) ALIGNMENT Permite realizar alineamientos completos utilizando algoritmos restrictivos que generan un árbol guía o algoritmos más rápidos. OUTPUT Formato del resultado (ALN, GCG, PHYLIP, PIR and GDE) OUTORDER Orden de las secuencias COLOR Muestra el alineamiento en colores (solo en formatos ALN or GCG) AVFPMILW RED Small (small+ hydrophobic (incl.aromatic -Y)) DE BLUE Acidic RHK MAGENTA Basic STYHCNGQ GREEN Hydroxyl + Amine + Basic - Q Others Gray Línea consenso "*" = residuos idénticos o conservados en todas las secuencias ":" = sustituciones conservadas "." = sustituciones semi-conservadas.
 ALIGNMENT. Permite realizar alineamientos completos utilizando algoritmos restrictivos que generan un árbol guía o algoritmos más rápidos. OUTPUT. Formato del resultado (ALN, GCG, PHYLIP, PIR and GDE) OUTORDER. Orden de las secuencias. COLOR. Muestra el alineamiento en colores (solo en formatos ALN or GCG) AVFPMILW. RED. Small (small+ hydrophobic (incl.aromatic -Y)) DE. BLUE. Acidic. RHK. MAGENTA. Basic. STYHCNGQ. GREEN. Hydroxyl + Amine + Basic - Q. Others. Gray. Línea consenso. * = residuos idénticos o conservados en todas las secuencias. : = sustituciones conservadas. . = sustituciones semi-conservadas.")
104
Clustal W (EBI)
")
105
Eliminar espacios entre secuencias
Formato secuencias para Clustal W Formato FASTA >FOSB_HUMAN P53539 homo sapiens (human). fosb protein MFQAFPGDYDSGSRCSSSPSAESQYLSSVDSFGSPPTAAASQECAGLGEMPGSFVPTVTA ITTSQDLQWLVQPTLISSMAQSQGQPLASQPPVVDPYDMPGTSYSTPGMSGYSSGGASGS GGPSTSGTTSGPGPARPARARPRRPREETLTPEEEEKRRVRRERNKLAAAKCRNRRRELT DRLQAETDQLEEEKAELESEIAELQKEKERLEFVLVAHKPGCKIPYEEGPGPGPLAEVRD LPGSAPAKEDGFSWLLPPPPPPPLPFQTSQDAPPNLTASLFTHSEVQVLGDPFPVVNPSY TSSFVLTCPEVSAFAGAQRTSGSDQPSDPLNSPSLLAL >FOSB_MOUSE P13346 mus musculus (mouse). fosb protein. ITTSQDLQWLVQPTLISSMAQSQGQPLASQPPAVDPYDMPGTSYSTPGLSAYSTGGASGS GGPSTSTTTSGPVSARPARARPRRPREETLTPEEEEKRRVRRERNKLAAAKCRNRRRELT LPGSTSAKEDGFGWLLPPPPPPPLPFQSSRDAPPNLTASLFTHSEVQVLGDPFPVVSPSY TSSFVLTCPEVSAFAGAQRTSGSEQPSDPLNSPSLLAL Eliminar espacios entre secuencias
. fosb protein. MFQAFPGDYDSGSRCSSSPSAESQYLSSVDSFGSPPTAAASQECAGLGEMPGSFVPTVTA. ITTSQDLQWLVQPTLISSMAQSQGQPLASQPPVVDPYDMPGTSYSTPGMSGYSSGGASGS. GGPSTSGTTSGPGPARPARARPRRPREETLTPEEEEKRRVRRERNKLAAAKCRNRRRELT. DRLQAETDQLEEEKAELESEIAELQKEKERLEFVLVAHKPGCKIPYEEGPGPGPLAEVRD. LPGSAPAKEDGFSWLLPPPPPPPLPFQTSQDAPPNLTASLFTHSEVQVLGDPFPVVNPSY. TSSFVLTCPEVSAFAGAQRTSGSDQPSDPLNSPSLLAL. >FOSB_MOUSE P13346 mus musculus (mouse). fosb protein. ITTSQDLQWLVQPTLISSMAQSQGQPLASQPPAVDPYDMPGTSYSTPGLSAYSTGGASGS. GGPSTSTTTSGPVSARPARARPRRPREETLTPEEEEKRRVRRERNKLAAAKCRNRRRELT. LPGSTSAKEDGFGWLLPPPPPPPLPFQSSRDAPPNLTASLFTHSEVQVLGDPFPVVSPSY. TSSFVLTCPEVSAFAGAQRTSGSEQPSDPLNSPSLLAL. Eliminar espacios entre secuencias.")
106
Resultados

107
Resultados JalView

108
Resultados Clustal W (.dnd)
")
109
Árboles Phylodendron Phylogenetic tree printer

110
Práctica ClustalW - Primers
Comparación múltiple Abrir una sesión Clustal W Fija los parámetros e introduce las secuencias usando el archivo múltiple al que hemos añadido todas las secuencias a alinear. Ejecutar Clustal W Visualizar los resultados Identificar y almacenar les regiones conservadas (primers) Diseño primers Realizar una traducción reversa de los primers en la Sequence Manipulation Suite de la Univ de Alberta Mediante las tablas de uso de codones, disminuir la degeneración de los primers adaptándolos al uso del Microorganismo problema Árboles Visualizar el archivo .dnd con Phylodendron (o TreeView local) Variar la topología del árbol enraizándolo a un outgroup arbitrario
 Diseño primers. Realizar una traducción reversa de los primers en la Sequence Manipulation Suite de la Univ de Alberta. Mediante las tablas de uso de codones, disminuir la degeneración de los primers adaptándolos al uso del Microorganismo problema. Árboles. Visualizar el archivo .dnd con Phylodendron (o TreeView local) Variar la topología del árbol enraizándolo a un outgroup arbitrario.")
Presentaciones similares











>")





.>")




