Descargar la presentación
La descarga está en progreso. Por favor, espere

1
Muestreo y Convergencia en calculos de Energia Libre de Interacciones Proteina-Ligando: El enlace de la tripenoxipiridina derivado a factor Xa y tripsina

2
Resumen Se estudia el muestreo y convergencia de los calculos de energia libre de un conjunto de 10 tripenoxipiridina derivados a dos serine proteases, factor Xa y tripsina Las mutaciones de un compuesto a otro involucran alrededor de 19 atomos, la creacion y aniquilacion de carga neta y varios modos alternativos de enlace. Se obtiene alta convergencia en los resultados (_+5-10 kJ/mol), se debe al uso de potenciales soft-core que facilitan la creacion y supresion de atomos, por una adecuada eleccion del camino de mutacion y por una escala de tiempo de un nanosegundo.
, se debe al uso de potenciales soft-core que facilitan la creacion y supresion de atomos, por una adecuada eleccion del camino de mutacion y por una escala de tiempo de un nanosegundo..")
3
Introduccion El calculo de EL es central porque todas las propiedades de equilibrio, como la fase, cte. de asociación-disociación, solubilidad, etc dependen de la diferencia de EL. La diferencia de EL esta relacionada a la probabilidad de encontrar a un sistema en un estado microscopico dado. El costo computacional es alto si se quiere calcular un promedio termico sobre las configuraciones microscopicas a un nivel atomico, para luego calcular la diferencia de EL entre dos estados.

4
Pero esto cambia si se usa potenciales intermedios modificados Se evaluara la afinidad de enlace relativa de un conjunto de 10 inhibidores a dos serine proteases, factor Xa y tripsina, que comparten secuencia y estructura homologa. Por este echo los sitios activos de muchos serine proteases son muy similares, y es importante que los inhibidores enlacen selectivamente al factor Xa y no a otra serine proteases, como la tripsina o trombina.

5
Los inhibidores analizados en este estudio tienen como molde la tripenoxipiridina, pero difieren en numero y tipo de sustituyentes, R1 y R2. Los inhibidores difieren unos de otros y para la transformacion de uno en otro involucran mutaciones. Entonces se requiere calculos adicionales y esto afecta la convergencia de los resultados.

6
Primero buscaremos la auto consistencia, explotando el echo que la EL es una funcion de estado asi que la diferencia de EL entre dos estados es independiente del camino escogido. Podemos escoger un camino reversible y evaluar le diferencia de EL de ida y de regreso.

7
Metodos Estudiaremos los complejos: factor Xa y tripsina con un conjunto de 10 inhibidores. Los sustituyentes R1 y R2 para cada inhibidor son mostrados en la tabla. Debido a que no tenemos data estructural del complejo, se uso como molde inicial la estructura cristalográfica del complejo: factor Xa y tripsina con 2,6- dipenoxipiridina.

8
La estructura de factor Xa es comprendida por dos cadenas polipeptidas. Sin embargo solamente se incluyo la cadena primaria el cual contiene lo sitios de enlace. La inicial estructura del complejo: factor Xa y tripsina con el inhibidor I1 fue construido por superponer los atomos de nitrogeno del grupo amino-metyl y los atomos de nitrogeno del grupo amidazolil del inhibidor sobre los correspondientes atomos en la apropiada estructura del molde.

9
La conformacion de I1 usado para este procedimiento fue tomado de una simulacion del inhibidor aislado en agua. La estructura del complejo: factor Xa y tripsina con los inhibidores I2-I10 fue derivado del complejo I1 por mutar los sustituyentes R1 y R2 durante el calculo de energia libre.

10
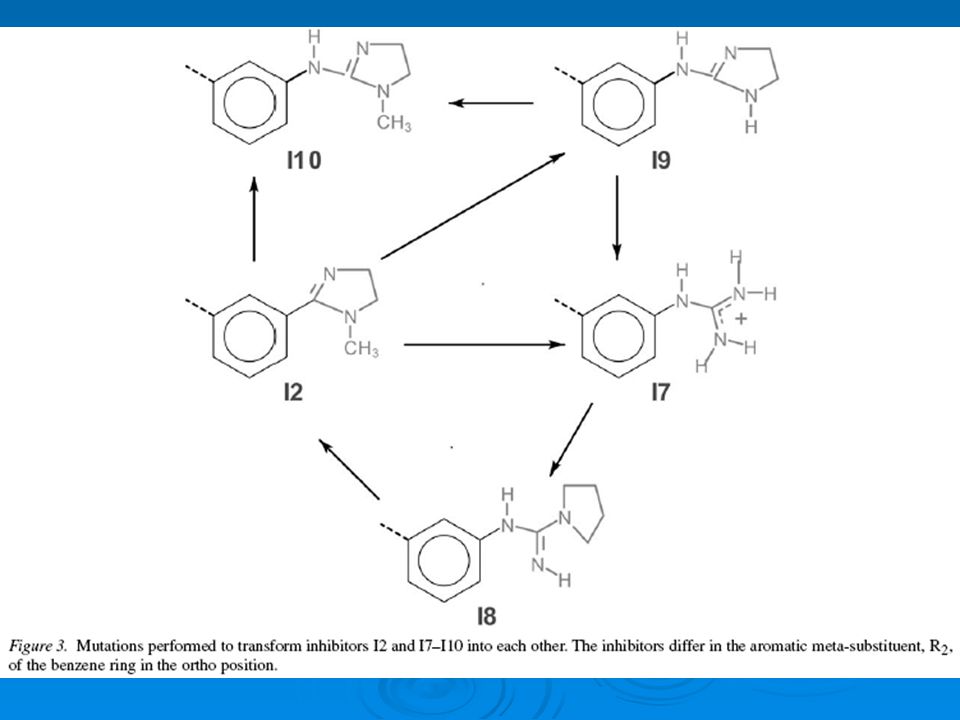
Mutaciones Las mutaciones fueron escogidas en orden a maximizar el numero de posibles ciclos cerrados mientras se minimiza el tamaño de la mutación misma. Como la EL de cualquier ciclo cerrado es cero entonces se puede comprobar la convergencia de los cálculos

12
Force Field La Gromos96 campo de fuerza fue usado para describir ambos: la proteína y el inhibidor. Cuando los parámetros no fueron compatibles para describir cierto tipo de interacción estos se obtuvieron por hacer un calculo de ab initio. Los parametros que no estaban disponibles incluido el potencial torsional entre el anillo penoxy y la piridina, y algunas cargas atomicas. Los calculos se hicieron con el metodo RHF con el programa gaussian94.

13
Las cargas atómicas fueron derivadas por hacer una escala de las cargas atómicas obtenidas por montar el potencial electrostático molecular RHF/6-31G de la correspondiente pequeña molécula orgánica, usando el metodo de Merz y Kollman. Los parametros adicionales derivados para el uso en lo calculos son presentados en la tabla.

14
Detalles computacionales Se utilizo el GROMACS para la simulación de MD en solvente explicito y bajo condiciones de frontera periódicas. El complejo proteína-inhibidor fue colocado en una caja octaédrica truncada que contiene aproximadamente 6300 moléculas de agua. Simulaciones del inhibidor libre en agua fue realizado en cajas que contienen aprox. 850 moléculas de agua. El rango limite de interacciones no enlazantes fue: 0.9nm-1.4nm. 0.9nm-1.4nm. Para la corrección del campo eléctrico mas allá del rango limite se utilizo una cte. Dieléctrica de ε RF =78.0 Para la corrección del campo eléctrico mas allá del rango limite se utilizo una cte. Dieléctrica de ε RF =78.0 El tiempo de paso usado para integrar la ecuación de movimiento fue 0.002 ps. La longitud de enlace y el ángulo de las moléculas de agua fueron restringidas usando el algoritmo de SETTLE mientras la longitud de enlace dentro la proteína fue restringido por el algoritmo de LINCS. El complejo Proteína-I1 fue equilibrado en 2 ns antes de calcular la EL. Para el caso del inhibidor libre en agua el tiempo de equilibrio fue 200 ps.

15
Cálculos de la Energía Libre

16
La EL de enlace,ΔG b, es el trabajo requerido para transferir al inhibidor de un estado libre en agua a un estado enlazado a la proteina. La relativa EL de enlace, Δ Δ G b X-Y, representa la diferencia en la EL de enlace entre el inhibidor X y el inhibidor Y y fue evaluado usando el ciclo termodinámico mostrado.

18
En el calculo de Δ G X-Y un inhibidor fue gradualmente mutado en otro inhibidor. En el caso donde no hubo directamente correspondencia entre los atomos en las dos moleculas se utilizo los atomos falsos. Solamente las interacciones enlazantes y no- enlazantes fueron mutadas durante los calculos. Las interacciones no-enlazantes entre el estado inicial y el estado final fueron interpoladas usando un potencial soft-core. Todas las mutaciones indicadas en la figura 2 y 3 fueron realizadas para los inhibidores en el complejo y los inhibidores en agua.

19
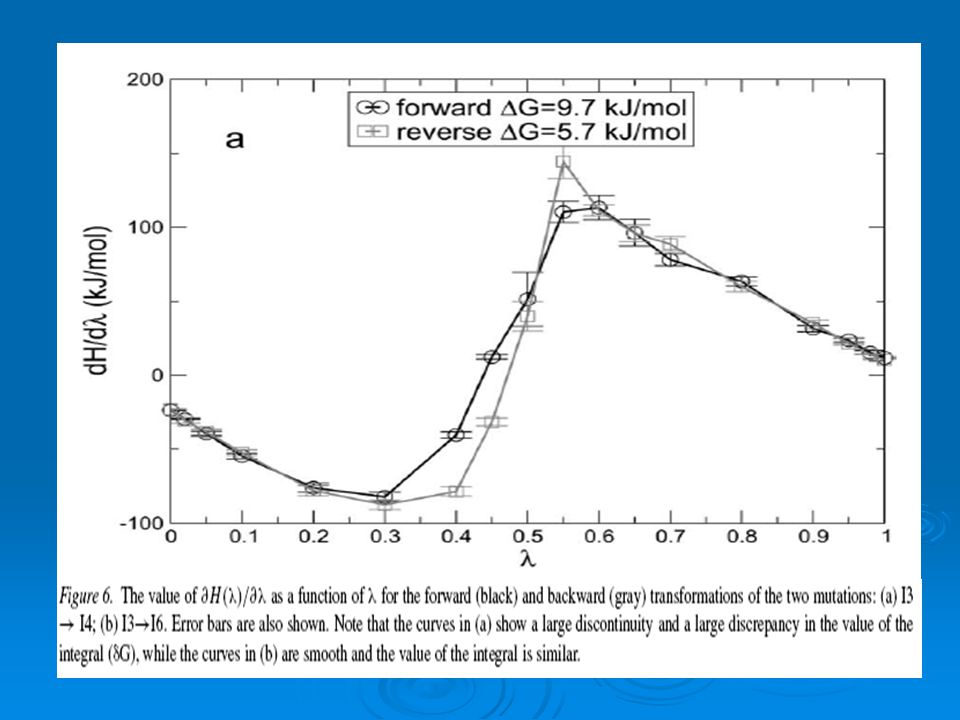
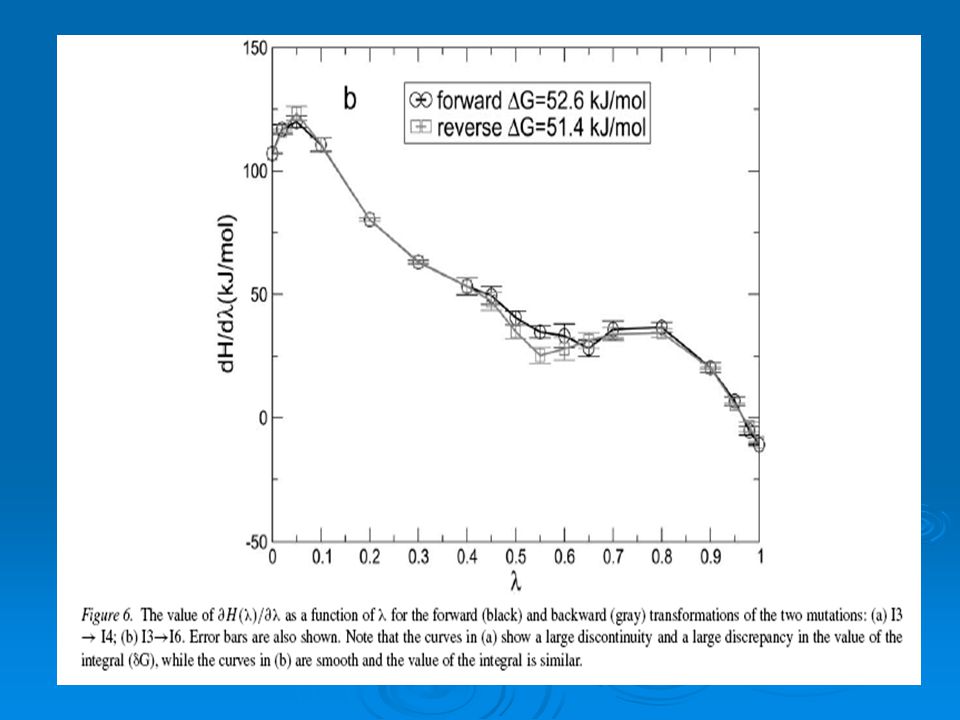
La EL fue evaluada usando 18 λ-puntos. El numero de λ-puntos fue incrementadas para casos donde la mutación genera una grande perturbación en el sistema in regiones donde la ecuación 1 como una función de λ exhibe una pronunciada discontinuidad. Se muestra en la fig.6 el valor de ∂H(λ)/∂λ de dos mutaciones. Para el caso de la mutación I3- I4 la curva es discontinua y para el caso de I3-I6 la curva es suave. El valor promedio de la derivada fue calculada en cada λ-punto y el resultado fue integrado usando el método del trapecio para obtener Δ G X-Y
/∂λ de dos mutaciones. Para el caso de la mutación I3- I4 la curva es discontinua y para el caso de I3-I6 la curva es suave. El valor promedio de la derivada fue calculada en cada λ-punto y el resultado fue integrado usando el método del trapecio para obtener Δ G X-Y.")
Presentaciones similares







>")


>")









