Descargar la presentación
La descarga está en progreso. Por favor, espere

1
SELECCION DE “TEMPLATES” Y ALINEAMIENTO

2
Energía X Nativa

3
Búsqueda homólogos con estructura conocida Selección homólogos de interés (miembros más cercanos, criterios adicionales) Obtención del alineamiento de la secuencia con los homólogos Cálculo del modelo Contraste del modelo Experimentación “Feed-back” con el modelo
 Obtención del alineamiento de la secuencia con los homólogos Cálculo del modelo Contraste del modelo Experimentación Feed-back con el modelo")
4
Búsqueda de homólogos de estructura conocida

5
SIMILITUD ENTRE SECUENCIAS INDICA SIMILITUD ENTRE ESTRUCTURAS Y FUNCION

6
HOMOLOGIA Y COMPARACION DE SECUENCIAS SIMILITUD ENTRE SECUENCIAS HOMOLOGIA (Mismo orígen evolutivo)
")
7
ALINEAMIENTO DE SECUENCIAS AGGVIIIQVG AGGVL-IQVG AGGVIIIQVGAGGVLIQVG

8
IMPORTANCIA ALINEAMIENTO Detección homólogos Construcción modelo estructural

9
Búsqueda de homólogos SECUENCIA INCOGNITA ATTVG...LMN BASE DE DATOS DE SECUENCIAS AGLM...WTKR TCGGLMN..HICG WRKCPGL...

10
COMPARACION SECUENCIAS Construir alineamiento óptimo Puntuar candidato

11
COMPARACION DE SECUENCIAS HOMOLOGIA ALTA:HOMOLOGIA ALTA: AWTRRATVHDGLMEDEFAA AWTRRATVHDGLCEDEFAA HOMOLOGIA BAJA: HOMOLOGIA BAJA: AWTRRAT AWTKLATAVVVFEGLCEDEWGG VHDGLMEDEFAA

12
Búsqueda de homólogos PUNTUACION SECUENCIAS BASE DATOS (Proteínas estructura conocida) LOCALIZACION CANDIDATO CON MAYOR PUNTUACION PREDICCION FUNCIONAL/ESTRUCTURAL POR ASOCIACION/”HERENCIA”
 LOCALIZACION CANDIDATO CON MAYOR PUNTUACION PREDICCION FUNCIONAL/ESTRUCTURAL POR ASOCIACION/ HERENCIA")
13
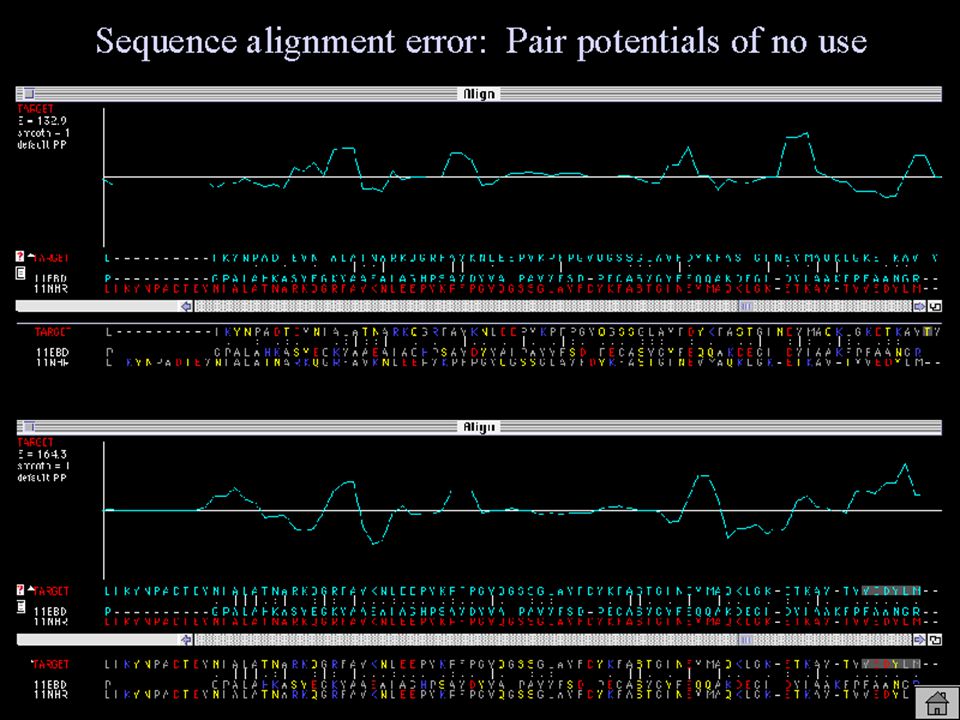
PROBLEMAS HABITUALES Solo hay familiares remotos: –BAJA SIMILITUD DE SECUENCIA –DIFICIL TENER BUENOS ALINEAMIENTOS

14
ALINEAMIENTO DE SECUENCIAS OBTENER EL ALINEAMIENTO OPTIMO NECESARIO: –METODO PARA PUNTUAR AMINOACIDOS COMPARADOS –METODO PARA CONSTRUIR LOS ALINEAMIENTOS

15
MATRIZ DE IDENTIDADES A C D...Y A 1 0 0...0 C 0 1 0...0..... Y 0 0.....1

17
NUMERO TOTAL DE ALINEAMIENTOS DOS SECUENCIAS DE LONGITUD N Y M: N + M N N = M = 1000 10 600

18
HERRAMIENTAS BASICAS MATRIZ COMPARACION AMINOACIDOS (Dayhoff, 1972) ALGORITMOS DE COMPARACION DE SECUENCIAS (Needleman & Wunsch, 1970).
 ALGORITMOS DE COMPARACION DE SECUENCIAS (Needleman & Wunsch, 1970).")
19
MATRIZ DE DAYHOFF log(fij/qi.qj) PUNTUACION DE LOS AMINOACIDOS ALINEADOS fij: frecuencia de mutación de residuo i al j qi, qj: frecuencia de los residuos i, j
 PUNTUACION DE LOS AMINOACIDOS ALINEADOS fij: frecuencia de mutación de residuo i al j qi, qj: frecuencia de los residuos i, j")
20
MATRIZ DE DAYHOFF REFLEJA LAS PROPIEDADES FISICO- QUIMICAS DE LOS AMINOACIDOS: propensidades de estructura secundaria hidrofobicidad volumen

21
MATRICES COMPARACION SECUENCIAS BLOSUM62 (Henikoff & Henikoff, 1992): derivada a partir de la comparación bloques de secuencias GONNET (Gonnet et al, 1992): alineamiento masivo de secuencias
: derivada a partir de la comparación bloques de secuencias GONNET (Gonnet et al, 1992): alineamiento masivo de secuencias")
22
ALGORITMO NEEDLEMAN & WUNSCH R G F Q R1 0 0 0 Y0 0 0 0 G0 1 0 0 Q0 0 0 1 R G F Q R3 1 1 0 Y2 1 1 0 G1 2 1 0 Q0 0 0 1 R_GFQ RYG_Q

23
GAPS (INSERCIONES/DELECIONES) LOCALIZADOS EN LOOPS
 LOCALIZADOS EN LOOPS")
24
? Candidato

25
GAPS (INSERCIONES/DELECIONES) ESQUEMAS DE PUNTUACION: –DEPENDIENDO DE ESTRUCTURA 2a –VALOR CONSTANTE –FUNCION LINEAL go + n. gl

26
PROGRAMACION DINAMICA VENTAJAS: PROPORCIONA UN ALINEAMIENTO REPRODUCIBLE Y OPTIMO DESVENTAJAS: ES LENTO

27
METODOS SUBOPTIMOS 10 – 100 MAS RAPIDOS PROPORCIONAN ALINEAMIENTOS SUBOPTIMOS BLAST, FASTA

28
BLAST BLAST (Altschul et al, 1990): localiza pequeños fragmentos comunes extenderlos hasta que la puntuación cae
: localiza pequeños fragmentos comunes extenderlos hasta que la puntuación cae")
29
BLAST RAPIDO, segundos EXPLORAR GENBANK, PDB FILTROS BAJA COMPLEJIDAD INDICES DE FIABILIDAD

30
ESTADISTICA INDICE DE REFERENCIA: E: número de falsos positivos esperado Búsquedas esporádicas: 0.01 – 0.001 Búsquedas masivas (anotación genoma) : 10 -6
 : 10 -6")
31
LIMITES COMPARACION SECUENCIAS EXISTENCIA DE PARENTESCOS INDETECTABLESEXISTENCIA DE PARENTESCOS INDETECTABLES PREDICCIONES ESTRUCTURALES DE BAJA CALIDAD EN MUCHOS CASOSPREDICCIONES ESTRUCTURALES DE BAJA CALIDAD EN MUCHOS CASOS

32
THE TWILIGHT ZONE IDENTIDAD INFERIOR AL 25 % SIMILITUD ESTRUCTURAL: HOMOLOGIA REMOTA Y ANALOGIA

33
THE TWILIGHT ZONE HOMOLOGIA REMOTA: ORIGEN EVOLUTIVO COMUN. E.G. HEMOGLOBINAS ANALOGIA: CONVERGENCIA ESTRUCTURAL. E.G. HEMOGLOBINA Y COLICINA

35
USO DE ALINEAMIENTOS MULTIPLES SIMILITUD BAJA, DOS SECUENCIAS:SIMILITUD BAJA, DOS SECUENCIAS: AVTTGLNMWTTAKRPGMDDFYTILLPGLMNCI GLFTAIDMHFFGRKPACEEYFTLVVDGLCNCI SIMILITUD BAJA, SECUENCIASMULTIPLES: SIMILITUD BAJA, SECUENCIASMULTIPLES: MPL ALTTGIDMWTTAKRPDMDDYYTIIIPGLMNCI MPTL AVTTGLNMWTTAKRPGMDDFYTILLPGLMNCI TMPTL GVTTGLNMYFTARRPGLDEFYTLVLRTLCMCL TMPTL GIFTDIDMHFYVKKPGLDEFFTLVLRTLCMAA MPTL AVTTGLNMWTTAKRPGMDDFYTILLPGLMNCI TMPTL GLFTALNMHFFGRKPACEEYFTLVVDGLCNCI

37
ALINEAMIENTOS MULTIPLES RESIDUOS CONSERVADOS: RELEVANTES PARA FUNCION O ESTRUCTURA PUNTUACION PONDERA LA CONSERVACION

38
PSI-BLAST BUSQUEDA UTILIZANDO ALINEAMIENTOS MULTIPLES: –BUSQUEDA BASE DE DATOS –CONSTRUCCION POSITION-SPECIFIC SCORE –ITERAR

39
PSI-BLAST PSI-BLAST NO ENCUENTRA LO QUE NO HAY EN LA PRIMERA BUSQUEDA BLAST DEFINIR E PARA LA INCLUSION DE SECUENCIAS (0.01)
")
40
THREADING/FOLD RECOGNITION ESTRATEGIA SIMILAR COMPARACION DE SECUENCIAS USO BASE DE DATOS ESTRUCTURAL DIFERENTE PUNTUACION CANDIDATOS

41
ATTWV....PRKSCT.......... 10.55.2>.......... CANDIDATO SELECCIONADO

42
COMPARACION SECUENCIA- ESTRUCTURA EVALUAR “EL GRADO DE AJUSTE DE LA SECUENCIA A LA ESTRUCTURA” UTILIZAR PROPIEDADES DIVERSAS: DISTANCIAS INTERRESIDUO, ESTRUCTURA SECUNDARIA, ETC

43
Selección templates: uso información adicional Proyecto: 2HNQ....HYTTWPDFGVP... CANDIDATO: 1YTS....HVGNWPDQTAV... Proyecto: 2HNQ.....HCSAGIGRS... CANDIDATO: 1YTS.....HSRAGVGRT................... 1YTS, 2HNQ: TIROSINA-FOSFATASA

44
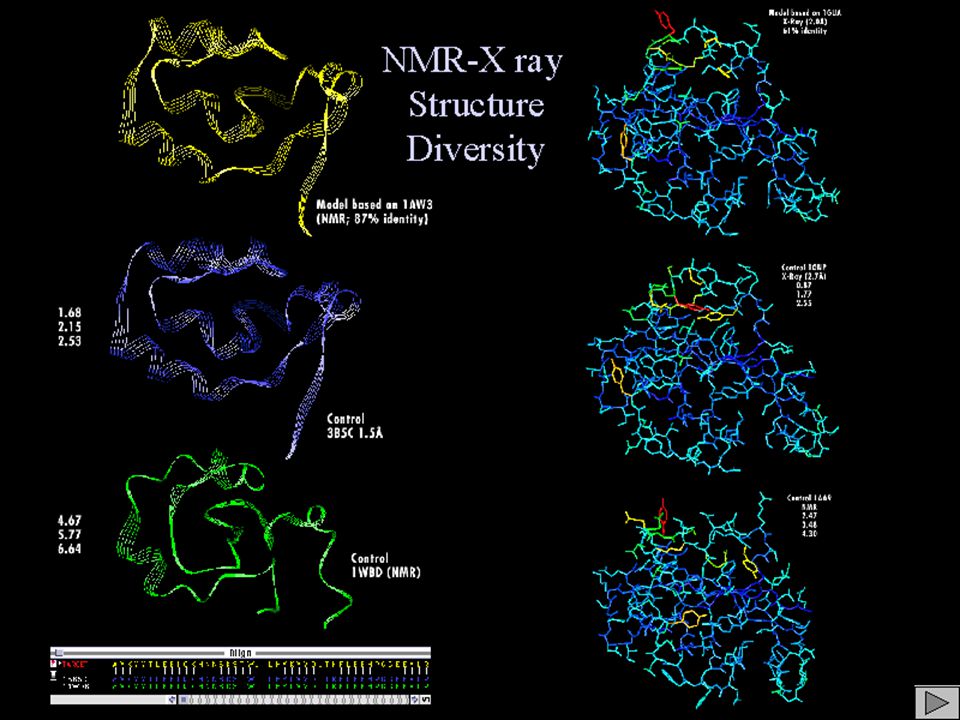
SELECCION “TEMPLATES” Varios candidatos, similitudes bajas (30% - 35 %) Bibliografía: sólo SSAO en H.Polymorpha y E.coli con TPQ en orientación correcta (modelos previos erróneos).
 Bibliografía: sólo SSAO en H.Polymorpha y E.coli con TPQ en orientación correcta (modelos previos erróneos).")
45
RESTRICCIONES CENTRO ACTIVO

46
SELECCION “TEMPLATES” Preferible X-ray sobre NMR Uso información experimental (e.g. centro activo) Preferible sin ligandos unidos, o generar dos modelos Vigilar la presencia de contactos en el cristal Preferible nativo sobre mutante
 Preferible sin ligandos unidos, o generar dos modelos Vigilar la presencia de contactos en el cristal Preferible nativo sobre mutante.")
47
EL ALINEAMIENTO

48
ALINEAMIENTOS SECUENCIA - CANDIDATO Entre candidatos estructurales

49
ALINEAMIENTO ENTRE CANDIDATOS Alineamientos estructurales, ya disponibles (HOMSTRAD), mediante software (SAP, Taylor & Orengo, 1989). Mejor pocos candidatos estructurales, y similares

50
ALINEAMIENTO TEMPLATES

51
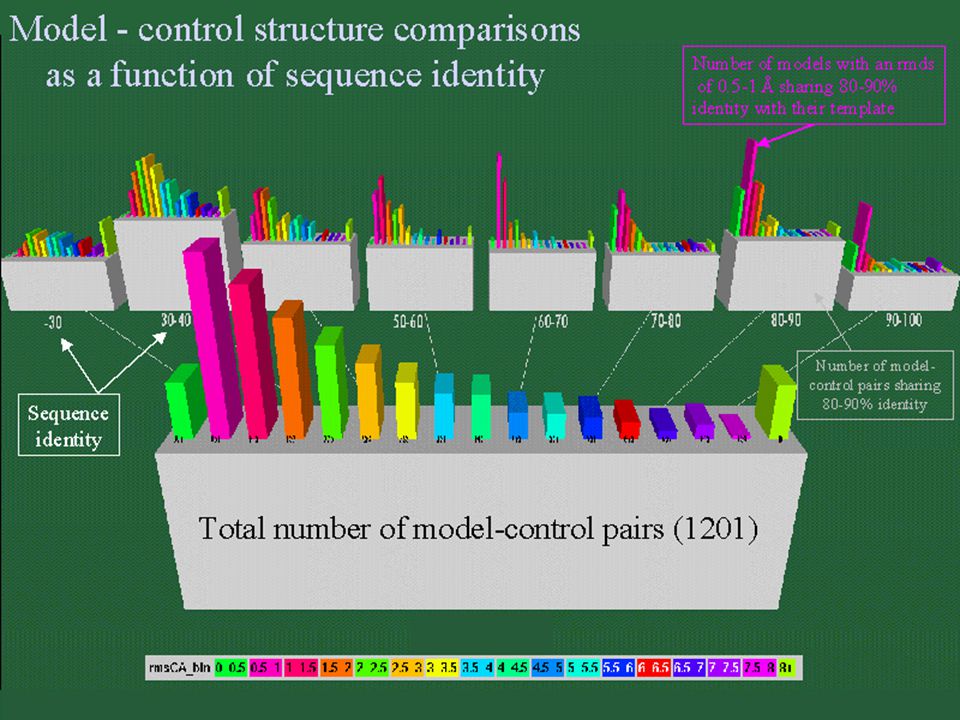
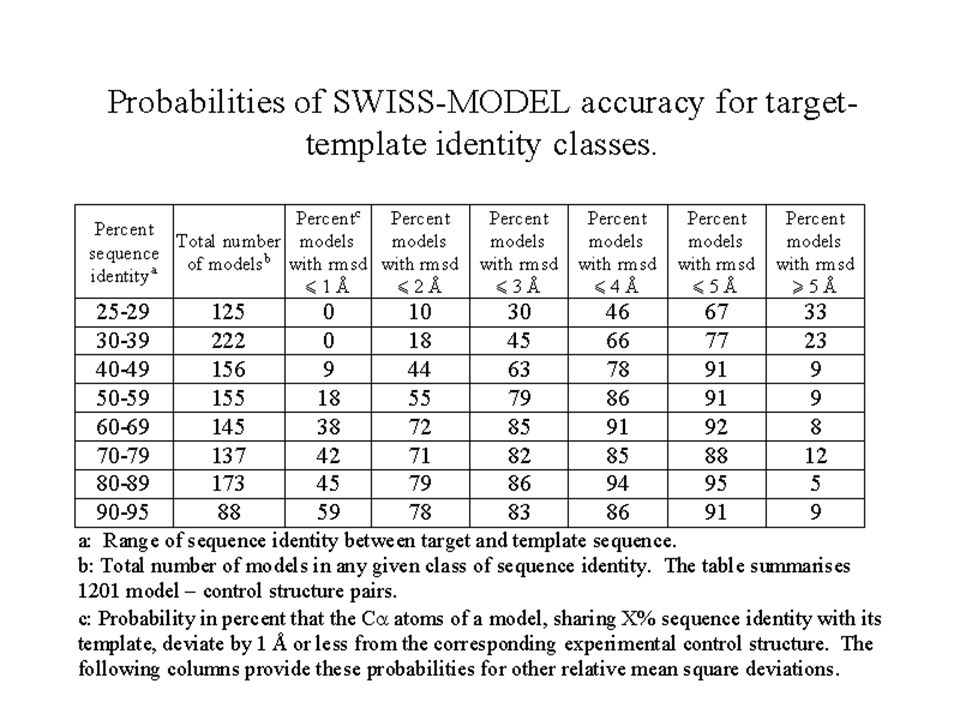
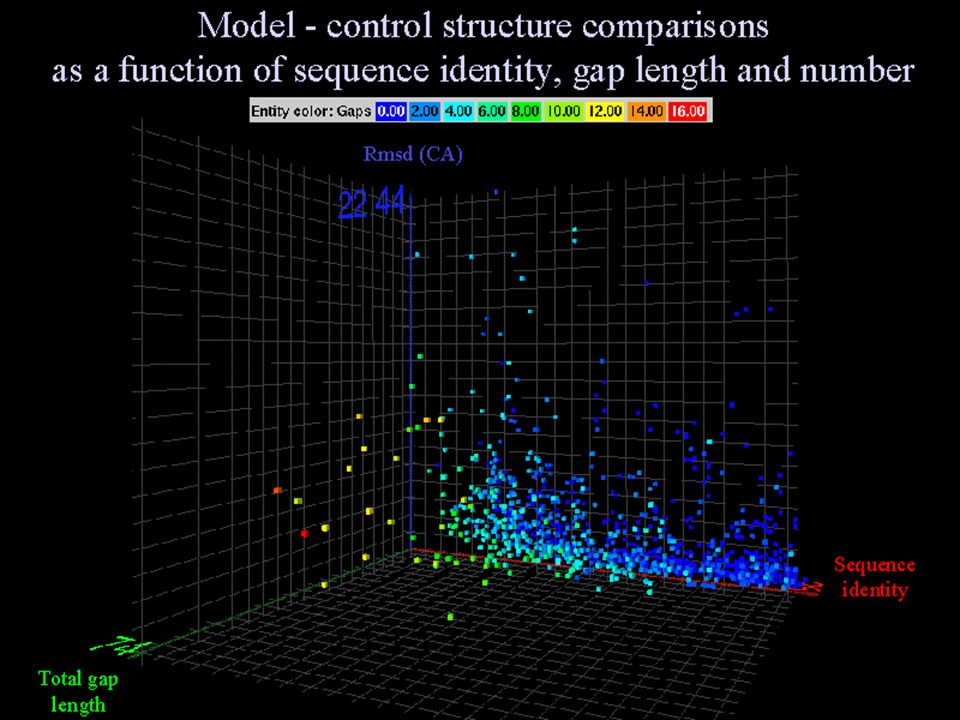
ALINEAMIENTO SECUENCIAS- CANDIDATOS Relación entre similitud estructural y % identidad Límite inferior del modelado: 30 % Porcentaje diseño de fármacos: > 70 %.

52
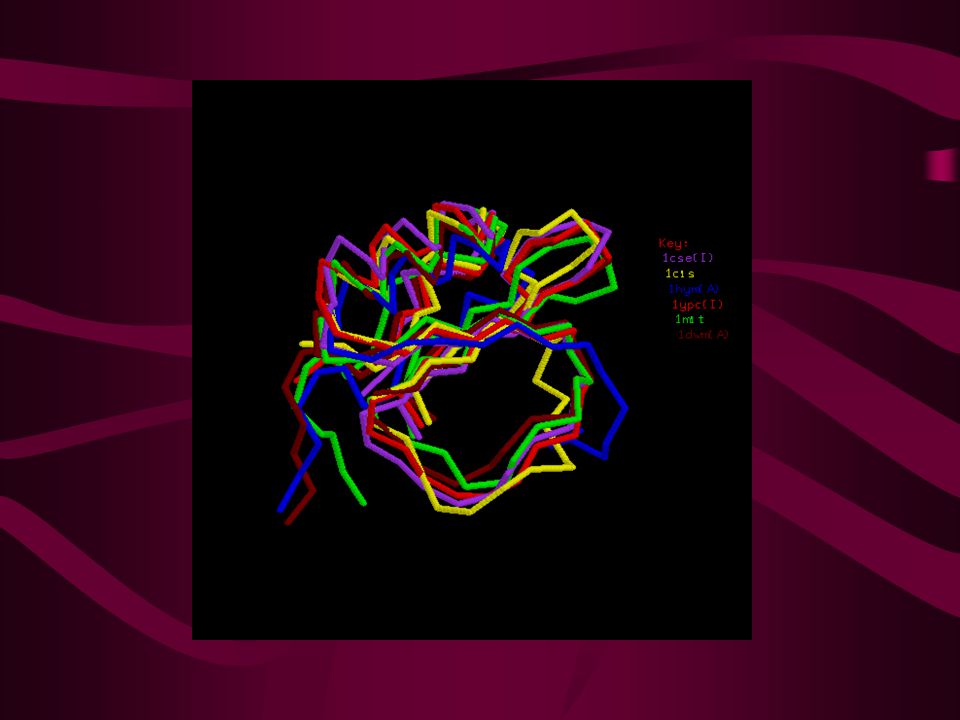
Similitud secuencia - estructura Rms % Ident.

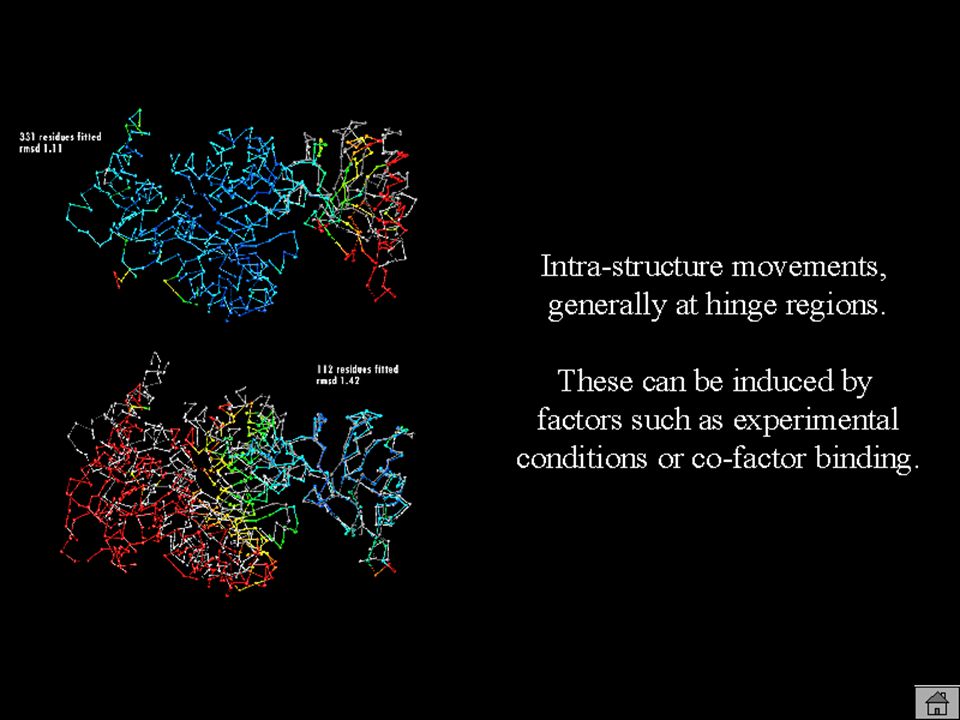
60
EL ALINEAMIENTO Baja similitud de secuencia => alineamiento baja calidad Número de resíduos alineados => limita calidad modelo (NW en GCG vs. BLAST)
.")
61
Energía X Nativa

62
Energía X Nativa

63
Energía X Nativa

64
Energía X Nativa

65
Energía X Nativa

66
ALINEAMIENTO Y MODELADO Nativa Modelo

67
CALIDAD ALINEAMIENTO Uso de potenciales fuerza media (PROSA) Uso de propiedades composicionales (GCG) Análisis de propiedades locales
 Uso de propiedades composicionales (GCG) Análisis de propiedades locales")
72
CALIDAD DEL ALINEAMIENTO Test globalTest global: comparar la secuencia con N pérmutas (N=1000). Calcular el Z-score resultante Si (alineamientos 100-200 aas): Z > 15 Ideal 5 < Z <= 15 70 % resíduos core bien alineados Z <= 5 Problemáticos
: Z > 15 Ideal 5 < Z <= % resíduos core bien alineados Z <= 5 Problemáticos.")
73
HHHHH--------HHHHHHHHHH-----HHHHHHHHHHHH-------HH LYLTIHSDHEGGNVSAHTSHLVGSALSDPYLSFAAAMNGLAGPLHGLAN LMVKVLDAVRGSPAINVAVHVFRKAADDTWEPFASGKTSESGELHGLTT EEEEEEE----EE----EEEEEEE-----EEEEEEEE-----EE----- Alineamiento: citrate synthase - transthyritin Z-score: 7.55

74
CALIDAD LOCAL Zonas mayor calidad: independientes de pequeños cambios en los parámetros del alineamiento (posible utilizando GCG) Zonas de mayor calidad: presentes en los alineamientos subóptimos
 Zonas de mayor calidad: presentes en los alineamientos subóptimos")
75
ALINEAMIENTOS SUBOPTIMOS Zonas comunes en los alineamientos subóptimos son las más fiables: alinear ALLIM vs. ALLM Sc. 7Sc.6 ALLIMALLIM ALL-MAL-LM

76
Alineamiento local Proyecto: 2HNQ....HYTTWPDFGVP... CANDIDATO: 1YTS....HVGNWPDQTAV... Proyecto: 2HNQ.....HCSAGIGRS... CANDIDATO: 1YTS.....HSRAGVGRT................... 1YTS, 2HNQ: TIROSINA-FOSFATASA

77
ALINEAMIENTO LOCAL Presencia de motivos PROSITE Adicionalmente: fijar estructura del motivo al construir el modelo.

78
ESTRATEGIA Utiliar siempre que sea posible: –Alineamientos estructurales para los templates (vigilar orígen: alineamientos sencillos o múltiples) –Alineamientos múltiples (e.g. Pfam) para alinear la secuencia a los templates –Análisis visual
 para alinear la secuencia a los templates –Análisis visual.")
79
Obtener alineamiento Pfam SSAO Obtener alineamiento HOMSTRAD 1spu, 1a2v de Pfam Eliminar 1spu, 1a2v de Pfam Alineamiento Homstrad con Pfam mediante CLUSTALW ALINEAMIENTO SSAO RATON CON LAS TEMPLATES

80
CONCLUSIONES: SENTIDO COMUN !! Analizar la calidad de los candidatos: ello decide el límite del modelado y del problema Examinar el alineamiento, y eventualmente generar varios modelos (buscar consistencia con datos experimentales)
.")
Presentaciones similares










>")











>")


>")
