Descargar la presentación
La descarga está en progreso. Por favor, espere

1
Fisiopatología Muscular
Dr. Pablo Monge Zeledón.

3
Fisiología muscular 40 – 45% del peso corporal
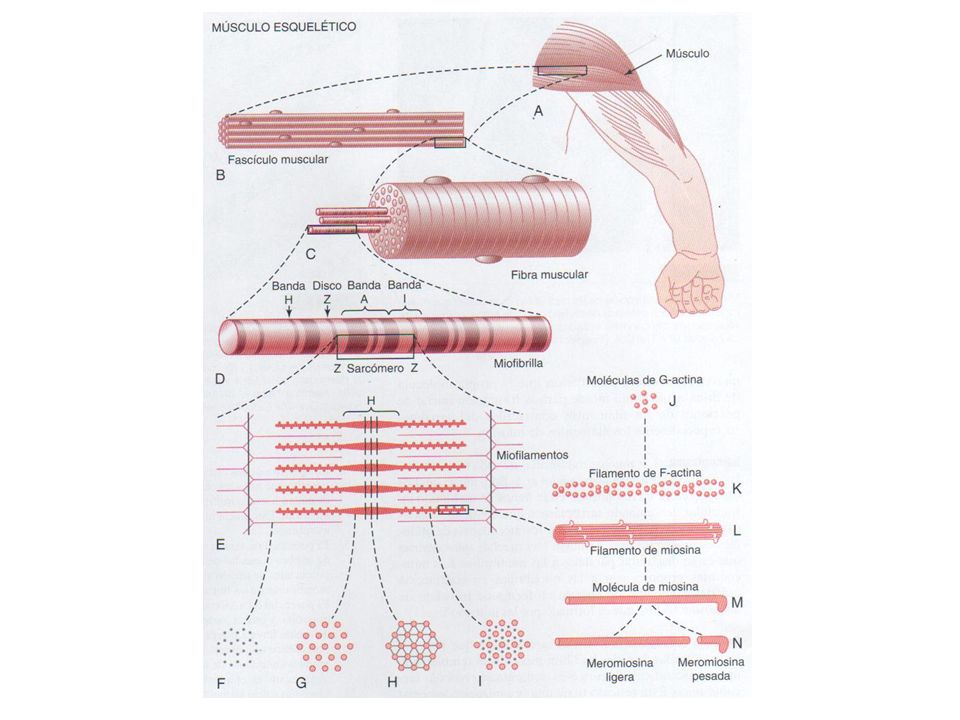
Macroscópicamente tiene forma fusiforme Rodeados por fascia Células gigantes multinucleadas (10 –100μm) Se organizan en fascículos: fibras tipo I, tipo IIa y tipo IIb. Miofibrillas de 1μm de diámetro. Filamentos de actina y miosina en paralelos que se engranan entre si.
 Se organizan en fascículos: fibras tipo I, tipo IIa y tipo IIb. Miofibrillas de 1μm de diámetro. Filamentos de actina y miosina en paralelos que se engranan entre si.")
5
Fisiología muscular

7
Fisiología muscular

9
Fibras musculares Tipo I Tipo II a Tipo II x Tipo II b
Tiempo Contracción Lento Moderado rápido Rápido Muy rápido Tamaño Pequeño Medio Grande Muy grandes Resistancia a la fatiga Alta Intermedia Baja Uso Aeróbico Anaeróbico largo plazo Anaeróbico corto plazo Maxima duración de uso Horas <30 minutos <5 minutos <1 minuto Fuerza Media Muy alta Mitocondrias Muchas Pocas Densidad Capilares Capacidad oxidativa Capacidad glicolítica Alto Almacén energético TG CK, glucógeno

12
Fibras musculares, miofibrillas y miofilamentos

13
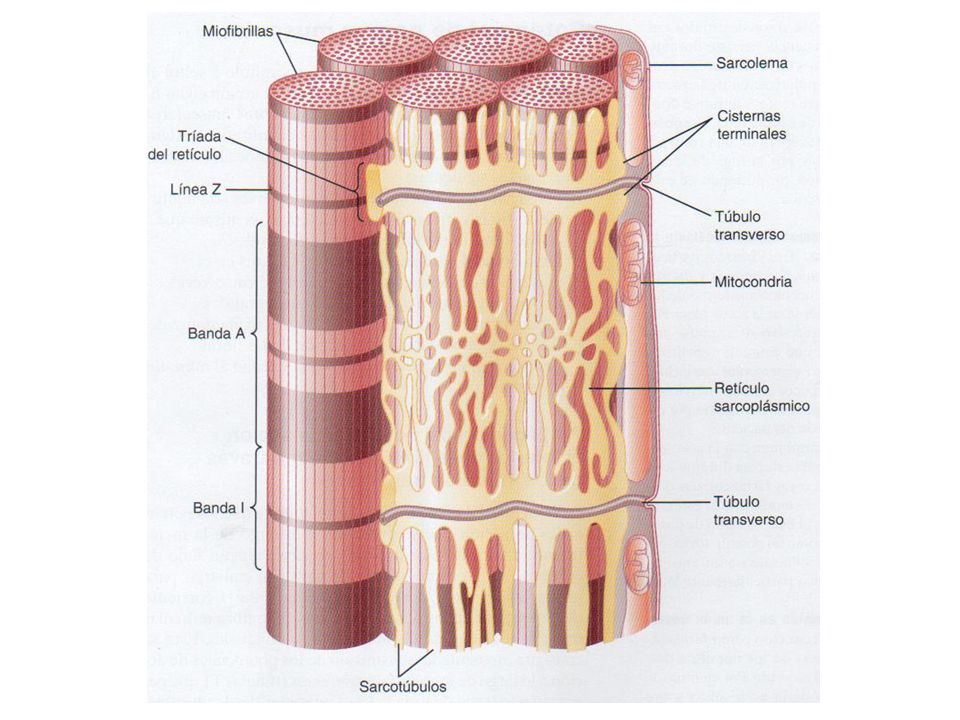
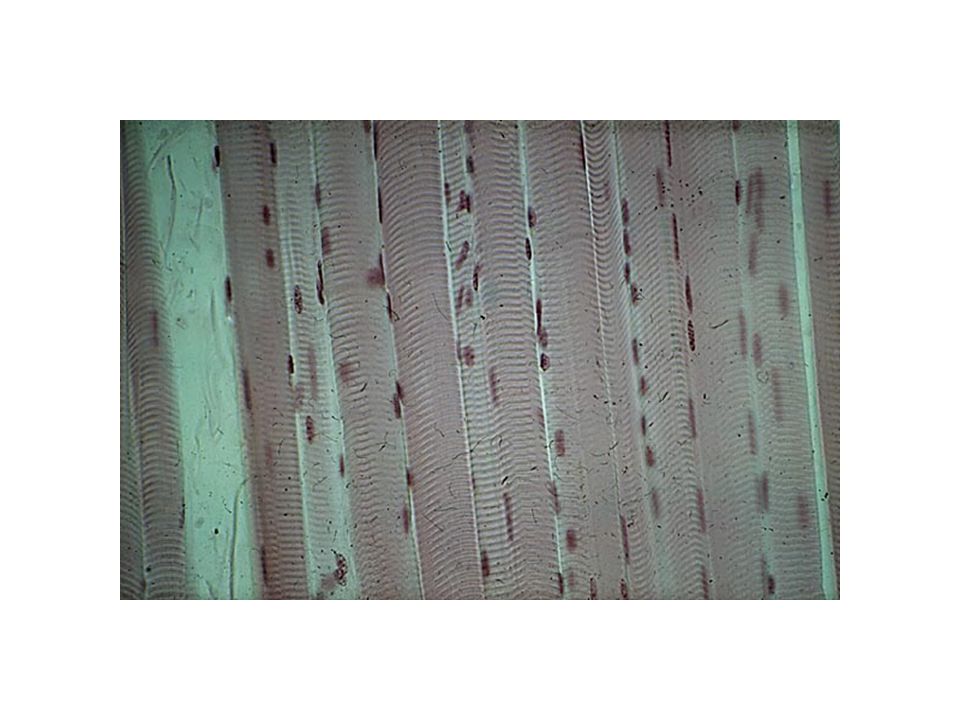
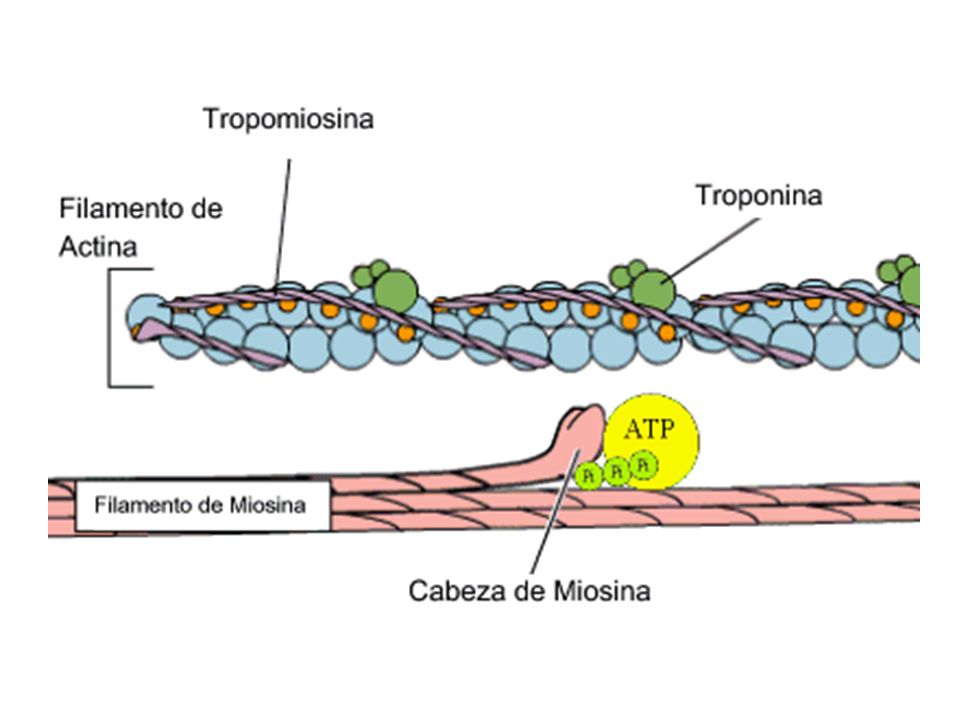
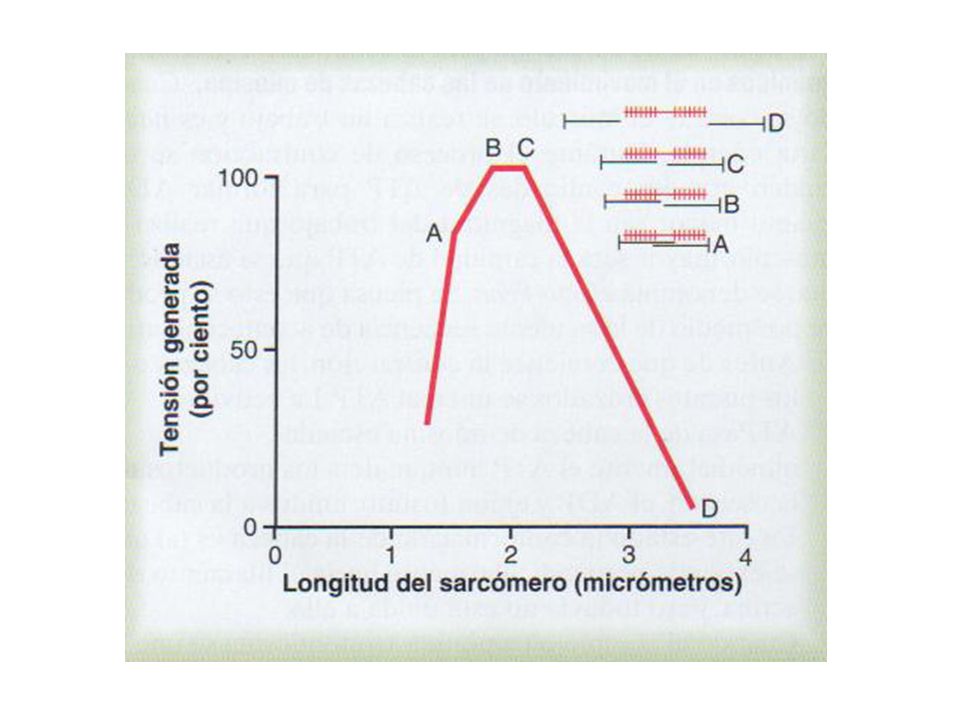
Sarcómero Línea Z Banda A: Miosina. Actina Banda I. Banda H. Línea M

14
Sarcómero

16
Sarcómero

18
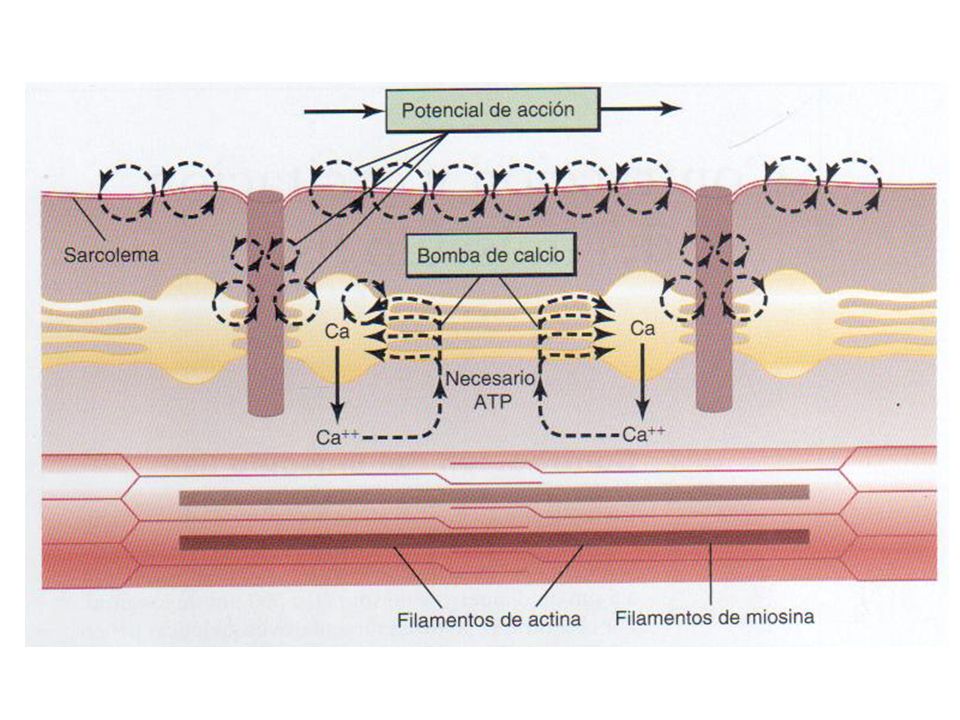
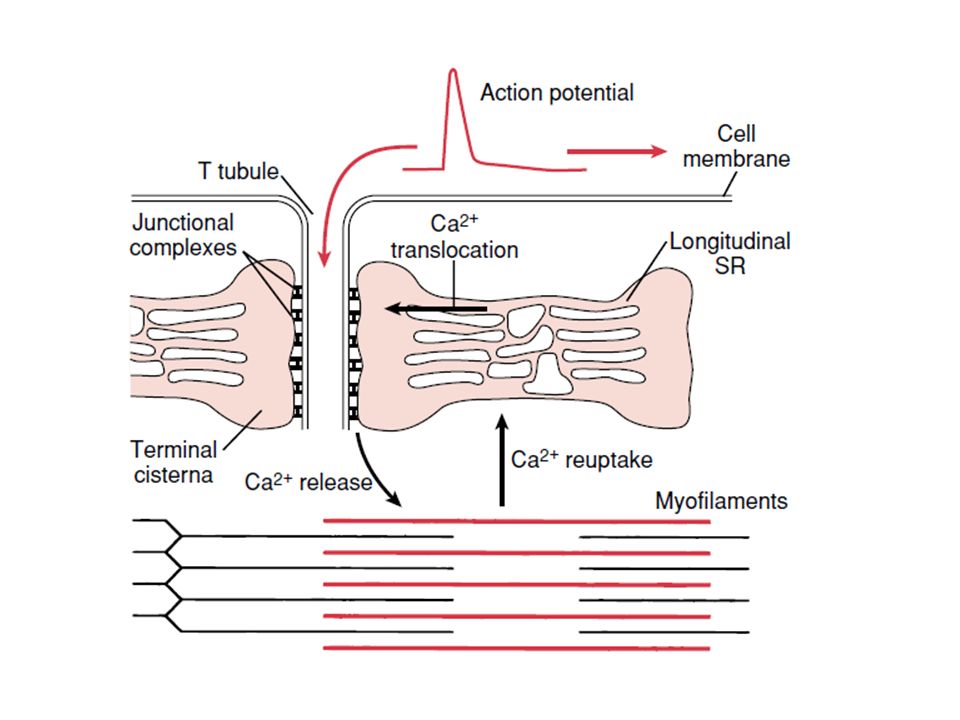
Fisiología muscular La contracción muscular se desencadena por impulsos procedentes del SN periférico. Potenciales de placa motora Despolarización del sarcolema Liberación de iones Ca++ Relajación Fosfatos ricos en energía: ATP, creatinafosfato. Fuente energética: glucosa, glucógeno

19
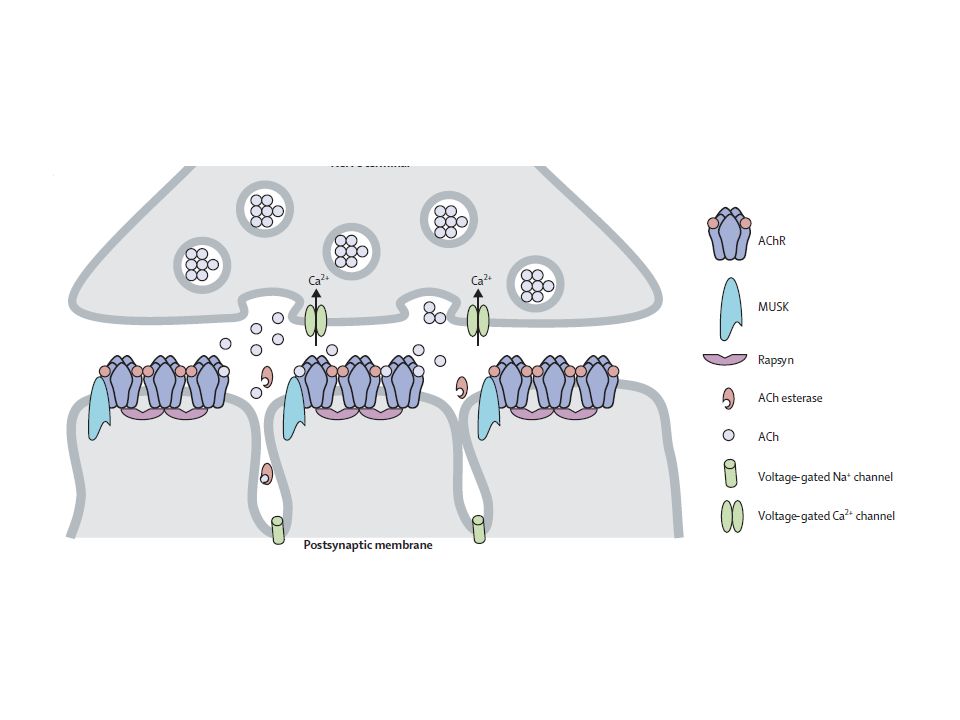
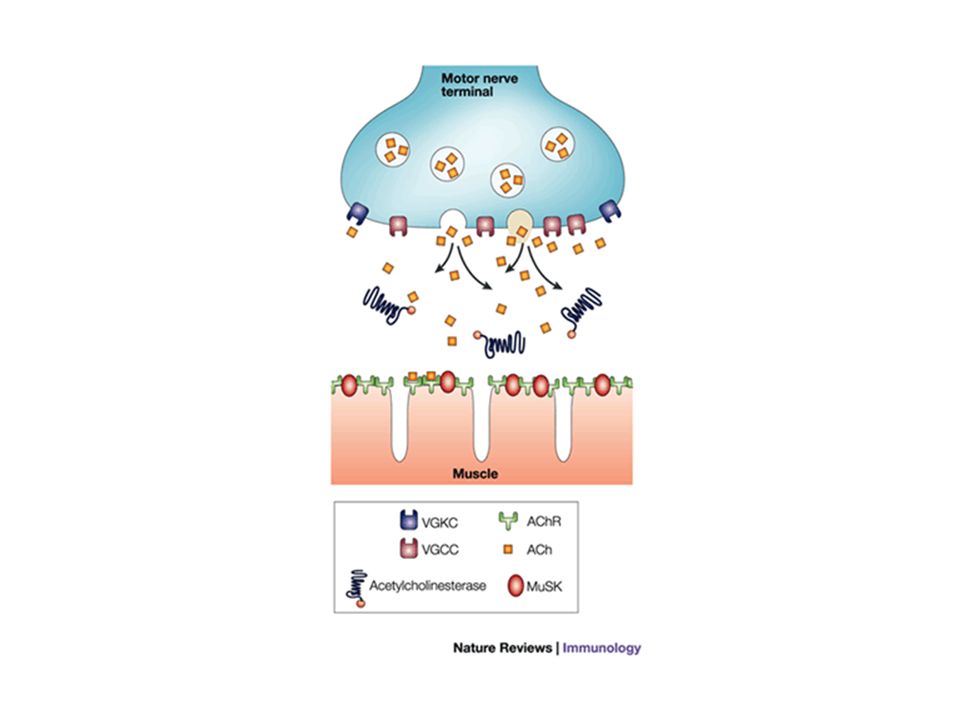
Placa motora

20
Placa motora

21
Placa motora

24
Bases moleculares de la contracción muscular

25
Bases moleculares de la contracción muscular

26
Bases moleculares de la contracción muscular

27
Bases moleculares de la contracción muscular
Eventos moleculares de la contracción muscular. En reposo, el ATP está unido a la cabeza de la miosina e hidrolizado, pero la energía de la reacción no puede ser liberada hasta que La cabeza de miosina pueda interactuar con la actina. La liberación de los productos de la hidrólisis es asociado con El golpe de poder Los puentes cruzados están ahora en el estado de rigor. La separación es posible cuando una nueva molécula de ATP se une a la cabeza de miosina. Esta es subsecuentemente hidrolizada A, actina; M, miosina; *, vínculo químico.

28
Enfermedades hereditarias
Distrofias musculares progresivas Miopatías congénitas con peculiaridades estructurales. Miopatías metabólicas hereditarias Enfermedades hereditarias de los canales iónicos

29
Distrofias musculares progresivas
Enfermedades con herencia distinta, que cursan con una degeneración progresiva de la musculatura esquelética. Según el tipo, la atrofia y la debilidad muscular predominan en los grandes grupos musculares proximales de la cintura pelviana o escapular Citoesqueleto: Sarcolema Distrofina (Responsable de la estabilidad de la membrana) Complejo sarcoglucano (Permite el anclaje) Matriz extracelular (Merosina y emerina)
 Complejo sarcoglucano (Permite el anclaje) Matriz extracelular (Merosina y emerina)")
30
Distrofias musculares progresivas

32
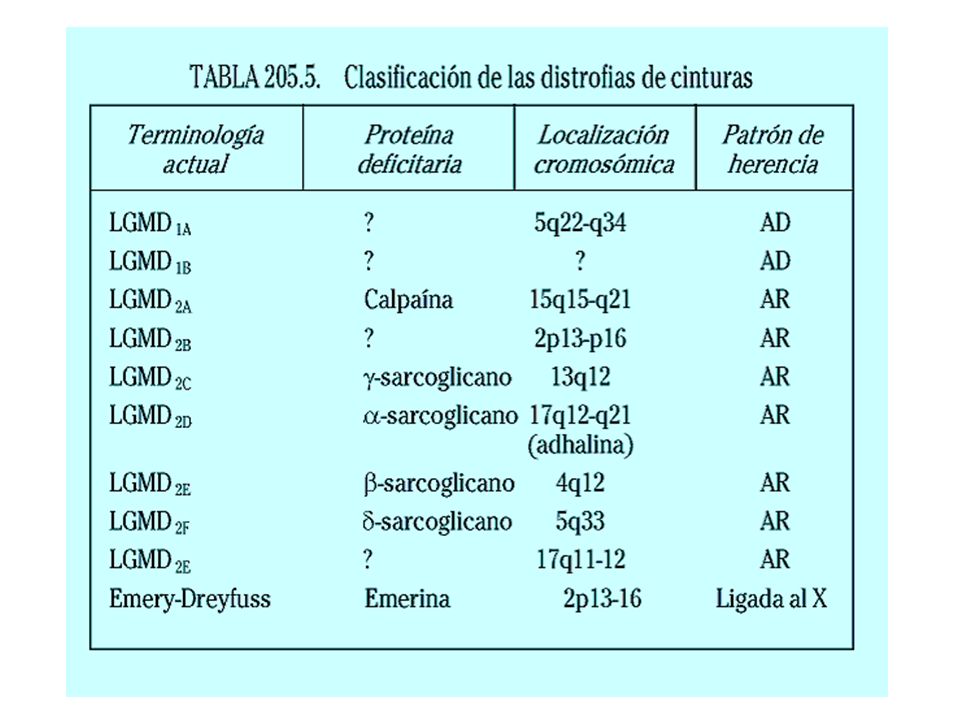
Distrofias musculares progresivas
Enfermedad Herencia Producto del gen Tipo Duchenne Recesiva ligada al cromosoma X (Xp21) Ausencia de distrofina Tipo Becker Recesiva ligada al cromosoma X Disminución de distrofina Facioescapulohumeral (Landouzy Dejerine) Autosómica dominante (4q35) Desconocido Cinturas (Limb-girdle muscular dystrophy LGMD) Autosómica recesiva Complejo sarcoglucano Congénitas Deficiencia de merosina Emery-Dreifuss Recesiva ligada al cromosoma X (Xq28) Deficiencia de emerina
 Ausencia de distrofina. Tipo Becker. Recesiva ligada al cromosoma X. Disminución de distrofina. Facioescapulohumeral. (Landouzy Dejerine) Autosómica dominante. (4q35) Desconocido. Cinturas. (Limb-girdle muscular dystrophy LGMD) Autosómica recesiva. Complejo sarcoglucano. Congénitas. Deficiencia de merosina. Emery-Dreifuss. Recesiva ligada al cromosoma X (Xq28) Deficiencia de emerina.")
33
Cromosoma X

34
Distrofias musculares progresivas
Inestabilidad de la membrana Salida del contenido de la célula muscular Elevación de la creatinafosfoquinasa (CPK) Degeneración del parénquima, sustituido por tejido adiposo y conjuntivo. Debilidad muscular Atrofia muscular
 Degeneración del parénquima, sustituido por tejido adiposo y conjuntivo. Debilidad muscular. Atrofia muscular.")
35
Duchenne Es la distrofia muscular más frecuente
Se transmite de forma recesiva y ligada al sexo La incidencia de esta enfermedad es alta 13-33 casos por habitantes Afecta a todos los países del mundo. Anomalía en la región p21 del cromosoma X.

36
Duchenne El producto proteico es la distrofina (427 kD)
Une la actina con DAG (dystrophin associated glycoproteins), que a su vez se unen a la matriz extracelular a través de la merosina. Las DAG se pueden agrupar en familias Distroglicanos Sarcoglicanos Sintrofinas La ausencia de algunas de estas proteínas facilita que la célula muscular sufra fenómenos de necrosis. Ello ocurre de manera especialmente manifiesta en ausencia de distrofina
, que a su vez se unen a la matriz extracelular a través de la merosina. Las DAG se pueden agrupar en familias. Distroglicanos. Sarcoglicanos. Sintrofinas. La ausencia de algunas de estas proteínas facilita que la célula muscular sufra fenómenos de necrosis. Ello ocurre de manera especialmente manifiesta en ausencia de distrofina.")
37
Distrofias musculares progresivas

38
Duchenne Se inicia en general antes de los 4 años
Continuas caídas al suelo al andar o al correr La cintura pelviana se afectan primer Dificultades para la extensión de rodillas y caderas. Afección de los músculos glúteos medios Marcha de pato. Se levantan con ayuda de las extremidades superiores, apoyando las manos en las rodillas para facilitar la elevación del tronco Signo de Gowers

39
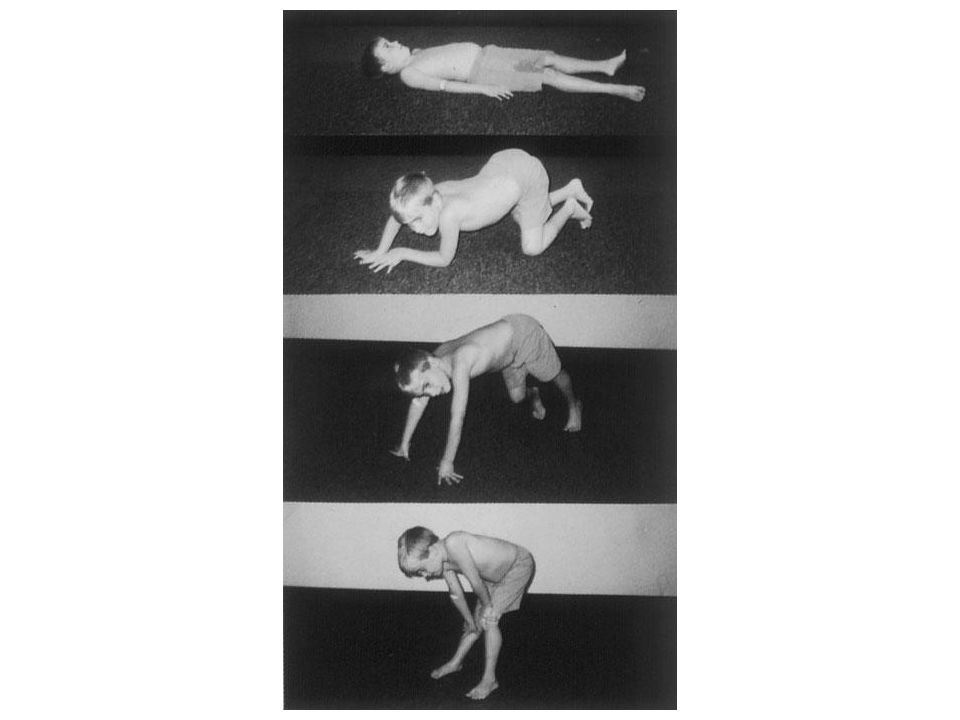
Signo de Gowers

41
Duchenne Pseudohipertrofia de las pantorrillas
Hiperlordosis lumbar al andar Marcha en "rey de comedia". La debilidad de los músculos que fijan las escápulas determina que éstas protruyan cuando el paciente levanta los brazos. La musculatura facial, ocular, bulbar y de las manos no se afecta hasta las fases finales de la enfermedad.

42
Marcha de Trendelenburg (balanceo de caderas)
Duchenne Pseudohipertrofia de pantorrillas Marcha de Trendelenburg (balanceo de caderas)
")
43
Duchenne Pierden la capacidad para deambular a los 10-15 años
Confinados en una silla de ruedas. ROT abolidos Contracturas musculares Posición con flexión y abducción de la cadera, flexión de la rodilla y pie en equinovaro. Suelen fallecer antes de los 25 años por insuficiencia respiratoria o cardíaca.

44
Duchenne Presentan cierto grado de lesión cardíaca.
Esta miocardiopatía cursa de forma asintomática o con trastornos del ritmo cardíaco También puede originar cuadros de insuficiencia cardíaca. La mayoría de estos pacientes presentan cierto déficit intelectual, más acusado al valorar la capacidad del habla. Los análisis de laboratorio suelen mostrar una gran elevación de las enzimas musculares séricas CPK, aldolasa, SGOT, DHL

45
Becker Los niveles de distrofina son casi normales pero la proteína es cualitativamente distinta. La clínica es semejante al Duchenne El inicio es alrededor de los 11 años El curso es más benigno. Deja de andar entre los 25 y 30 años Fallecen después de la quinta década de la vida. Pseudohipertrofia de pantorrillas y contracturas. Afección cardíaca menos frecuente Capacidad intelectual suele mantenerse normal. La CPK se encuentra elevada

46
Distrofia muscular facioescapulohumeral
Landouzy-Déjerine Afecta sobre todo los hombros y la cara Autosómica dominante Cromosoma 4q35 Inicio entre los 6 y los 20 años Dificultad para elevar los brazos o aparición de una escápula alada. Debilidad de la musculatura facial con dificultad para cerrar firmemente los ojos, silbar o succionar. Se afectan siempre la facies y la cintura escapular La afección de la cintura pelviana es muy posterior.

47
Distrofias musculares progresivas
Distrofia Facioescapulohumeral (Escapula alada)
")
48
Distrofia muscular facioescapulohumeral
No se observan seudohipertrofias ni contracturas. La función mental es normal Las lesiones cardíacas son raras La CPK y otras enzimas musculares son normales o están ligeramente elevadas.

49
Distrofia muscular de la cinturas
(LGMD) Debidas a defectos de las DAG (sarcoglicanos) o de la calpaína Emery-Dreifuss Herencia ligada al sexo Xq28 Emerina Transporte de vesículas transmembrana. Tríada diagnóstica Debilidad progresiva escapulo-húmero-peroneal, Contracturas precoces Cardiomiopatía con bloqueos de conducción
 Debidas a defectos de las DAG (sarcoglicanos) o de la calpaína. Emery-Dreifuss. Herencia ligada al sexo. Xq28. Emerina. Transporte de vesículas transmembrana. Tríada diagnóstica. Debilidad progresiva escapulo-húmero-peroneal, Contracturas precoces. Cardiomiopatía con bloqueos de conducción.")
51
Miopatías congénitas con peculiaridades estructurales
Enfermedades hereditarias autosómicas, en menos casos ligadas al cromosoma X, que suelen manifestarse ya en el nacimiento y su curso es relativamente benigno. Enfermedad del core central Miopatía nemalina Miopatía centronuclear Miopatía miotubular asociada al cromosoma X

52
Miopatías congénitas con peculiaridades estructurales
Enfermedad Herencia Producto del gen Enfermedad del core central. Autosómica dominante (19q) Receptor de rianodina Miopatía nemalínica Autosómica dominante (1q21-23) α-tropomiosina Miopatía centronuclear Autosómica dominante Desconocido Miopatía miotubular asociada al cromosoma X Recesiva ligada al cromosoma X (Xq18) Miotubulina
 Receptor de rianodina. Miopatía nemalínica. Autosómica dominante (1q21-23) α-tropomiosina. Miopatía centronuclear. Autosómica dominante. Desconocido. Miopatía miotubular asociada al cromosoma X. Recesiva ligada al cromosoma X (Xq18) Miotubulina.")
53
Miopatías congénitas con peculiaridades estructurales
Enfermedad del core central Anomalía redondeada de las fibrillas, que se produce en multiples fibras. Miopatía nemalínica (Nema = hilo) Bastones en fila a modo de red, formados por material de la línea Z condensado (Tricrómico de Gomori) Miopatía centronuclear Localización central de los núcleos de las fibras musculares Miopatía miotubular asociada al cromosoma X Similar a la anterior más estructuras similares a miotúbulos
 Bastones en fila a modo de red, formados por material de la línea Z condensado (Tricrómico de Gomori) Miopatía centronuclear. Localización central de los núcleos de las fibras musculares. Miopatía miotubular asociada al cromosoma X. Similar a la anterior más estructuras similares a miotúbulos.")
54
Miopatías metabólicas hereditarias
Grupo de enfermedades hereditarias autosómicas recesivas. Se caracteriza por una deficiencia enzimática determinada que afecta el metabolismo de: Hidratos de carbono Lípidos Las mitocondrias Las purinas

55
Miopatías metabólicas hereditarias
Hidratos de carbono Enfermedades por almacenamiento del glucógeno 9 enfermedades Tienen en común defectos en la síntesis de glucógeno, glucogenolisis o glucolisis Intolerancia al ejercicio Dolor Fatiga Rigidez Debilidad Calambres intensos Rabdomiolisis asociada al ejercicio

57
Miopatías metabólicas hereditarias
Glucogenosis de tipo II (POMPE) Déficit maltasa ácida (alfa glucosidasa ácida lisosómica) Autosómico recesivo (17q25.2) Cardiomegalia e insuficiencia cardíaca Retardo mental Afección musculoesquelética leve Glucogenosis de tipo III (FORBES) Déficit enzima desramificante (amilo-alfa-1,6 glucosidasa) Hepatomegalia. Hipotonía muscular Miocardiopatía Autosómico recesivo
 Déficit maltasa ácida (alfa glucosidasa ácida lisosómica) Autosómico recesivo (17q25.2) Cardiomegalia e insuficiencia cardíaca. Retardo mental. Afección musculoesquelética leve. Glucogenosis de tipo III (FORBES) Déficit enzima desramificante (amilo-alfa-1,6 glucosidasa) Hepatomegalia. Hipotonía muscular. Miocardiopatía. Autosómico recesivo.")
58
Miopatías metabólicas hereditarias
Glucogenosis de tipo IV (ANDERSEN) Déficit enzima ramificante Glucogenosis de tipo V (McARDLE) Déficit miofosforilasa. Suele comenzar después de los 20 años Calambres asociados al esfuerzo Rabdomiolisis ocasional (Mioglobinuria) Autosómico recesivo Glucogenosis de tipo VII (TARUI) Déficit de fosfofructoquinasa Similar al McArdle
 Déficit enzima ramificante. Glucogenosis de tipo V (McARDLE) Déficit miofosforilasa. Suele comenzar después de los 20 años. Calambres asociados al esfuerzo. Rabdomiolisis ocasional (Mioglobinuria) Autosómico recesivo. Glucogenosis de tipo VII (TARUI) Déficit de fosfofructoquinasa. Similar al McArdle.")
59
Miopatías metabólicas hereditarias
Lípidos Déficit de carnitina muscular Debilidad muscular en la niñez Asociado con mialgias, mioglobinuria, cardiomiopatía Aumento de CPK Aumento de lípidos en los músculos Déficit de Carnitina-palmitoil-transferasa Afecta tipicamente hombres Mialgias, calambres, rigidez y mioglobinuria. CPK normal.

60
Miopatías metabólicas hereditarias
Miopatías mitocondriales Grupo heterogéneo Anormalidades en el número, tamaño o estructura de la mitocondria (25 tipos) Puede asociarse con oftalmoplegia Kearns Sayre (Bloqueo AV completo) Intolerancia al ejercicio o debilidad muscular progresiva
 Puede asociarse con oftalmoplegia. Kearns Sayre (Bloqueo AV completo) Intolerancia al ejercicio o debilidad muscular progresiva.")
61
Enfermedades de los canales iónicos hereditarias
Se caracterizan por la alteración de un canal iónico determinado. Miotonías (Relajación muscular retrasada) Parálisis periódicas Canales de cloro Miotonía congénita dominante THOMSEN Miotonía congénita recesiva BECKER Canales de sodio Parálisis periódica hiperkalémica (hiperpotasémica) Paramiotonía congénita EULENBURG Canales de calcio Parálisis periódica hipokalémica
 Parálisis periódicas. Canales de cloro. Miotonía congénita dominante THOMSEN. Miotonía congénita recesiva BECKER. Canales de sodio. Parálisis periódica hiperkalémica (hiperpotasémica) Paramiotonía congénita EULENBURG. Canales de calcio. Parálisis periódica hipokalémica.")
63
Síndromes miotónicos Distrofia miotónica de Steinert
Miotonía Atrofia muscular Cambios distróficos de tejidos no musculares Cristalino, testículos, glándulas endocrinas, piel y encéfalo. Autosómica dominante Cromosoma19q13.3 Regula la síntesis de una proteincinasa muscular

64
Síndromes miotónicos Distrofia miotónica de Steinert
Inicio en la tercera década de la vida Debilidad muscular Elevador de los párpados Maseteros Esternocleidomastoideo Antebrazos Manos Músculos pretibiales.

65
Síndromes miotónicos Miotonía congénita autosómica (Thomsen)
Síntomas hasta los 10 años de edad Fenómeno miotónico más generalizado No se asocia a cardiopatía ni déficit intelectual. Rigidez generalizada Se acentúa con el reposo y en los ambientes fríos Mejora gradualmente con el ejercicio. Tienden a mejorar de forma progresiva Defecto en los canales del cloro

66
Síndromes miotónicos Miotonía congénita autosómica recesiva (Becker)
Más común en Alemania que en el resto de países. Más invalidante que la miotonía de Thomsen Inicio más tardío (12 años) Empieza en las piernas, afectando después los brazos y la cara. Defectos en los canales de cloro.
 Empieza en las piernas, afectando después los brazos y la cara. Defectos en los canales de cloro.")
67
Síndromes miotónicos Paramiotonía congénita (Eulenberg)
Miotonía tras exposiciones al frío Asociado a pérdidas transitorias de la fuerza muscular. La actividad muscular agrava la miotonía (miotonía paradójica), al contrario de lo que sucede con el fenómeno miotónico clásico. Defecto involucra la subunidad a del canal del sodio muscular.
, al contrario de lo que sucede con el fenómeno miotónico clásico. Defecto involucra la subunidad a del canal del sodio muscular.")
68
Síndromes miotónicos Parálisis periódica hiperpotasémica
Autosómica dominante Defecto en la subunidad a del canal del sodio Aparece en edades precoces Ataques duran entre 20 min y varias horas Extremidades inferiores Pueden desencadenarse por reposo tras un ejercicio, por frío, ayuno, gestación o administración de potasio Acompañando a la crisis se observan fenómenos miotónicos

69
Síndromes miotónicos Miotonía condrodistrófica (Schwartz-Jampel)
Autosómica recesiva Propia de la infancia Miotonía Retraso del crecimiento Atrofia muscular Deformidades esqueléticas.

70
Enfermedades de los canales iónicos hereditarias

71
Parálisis periódica Parálisis periódicas familiares hipopotasémicas
Autosómica dominante Cromosoma 1q31-32 Subunidad a del canal del calcio muscular. Inicia entre los 10 y los 20 años Episodios agudos de parálisis Extremidades inferiores Primero los músculos proximales ROTs disminuidos o abolidos durante la crisis

72
Parálisis periódica Parálisis periódicas familiares hipopotasémicas
Cuadro dura entre 12 y 48 h Desencadenadas por el frío, el reposo tras un ejercicio o una ingesta rica en hidratos de carbono y/o sodio. Potasio sérico durante las crisis es siempre bajo Enzimas musculares séricas son normales

73
Miopatías secundarias (Adquiridas)
")
75
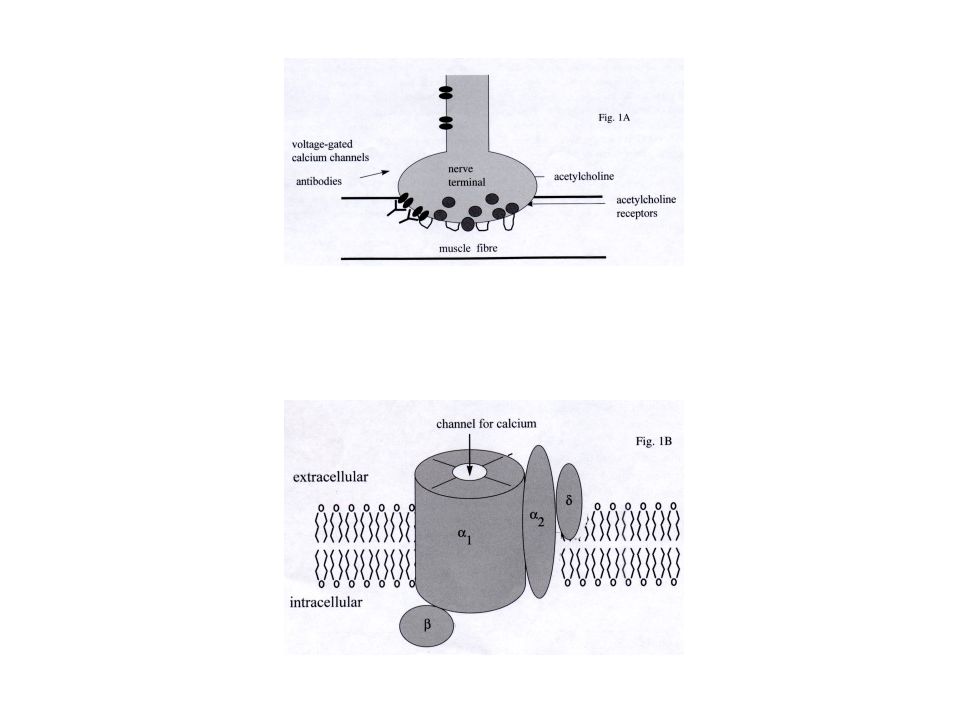
Miastenia gravis Enfermedad de la unión neuromuscular
Naturaleza autoinmune Destrucción específica, mediada por Ac, de los receptores de acetilcolina de la membrana postsináptica de la placa motora (90% de los pacientes) Aparición de debilidad muscular Tras una actividad prolongada Con recuperación después de un período de inactividad o la administración de fármacos anticolinesterásicos.
 Aparición de debilidad muscular. Tras una actividad prolongada. Con recuperación después de un período de inactividad o la administración de fármacos anticolinesterásicos.")
76
Miastenia gravis Etiología no clara Predisposición genética
Asociación con antígenos HLA A1, B8, DR3 5% presentan hipertiroidismo concomitante 30% con pariente materno con un trastorno autoinmune Presencia con mayor frecuencia de otras enfermedades autoinmunes: Artritis reumatoide, LES, polimiositis Hiperplasia tímica (65%) Timoma ( %)
 Timoma ( %)")
77
Miastenia gravis Simplificación de la región postsináptica
Ausencia de hendiduras sinápticas O espaciadas, poco profundas y anchas Vesículas presinápticas normales Colecciones dispersas de linfocitos T

78
Miastenia gravis

79
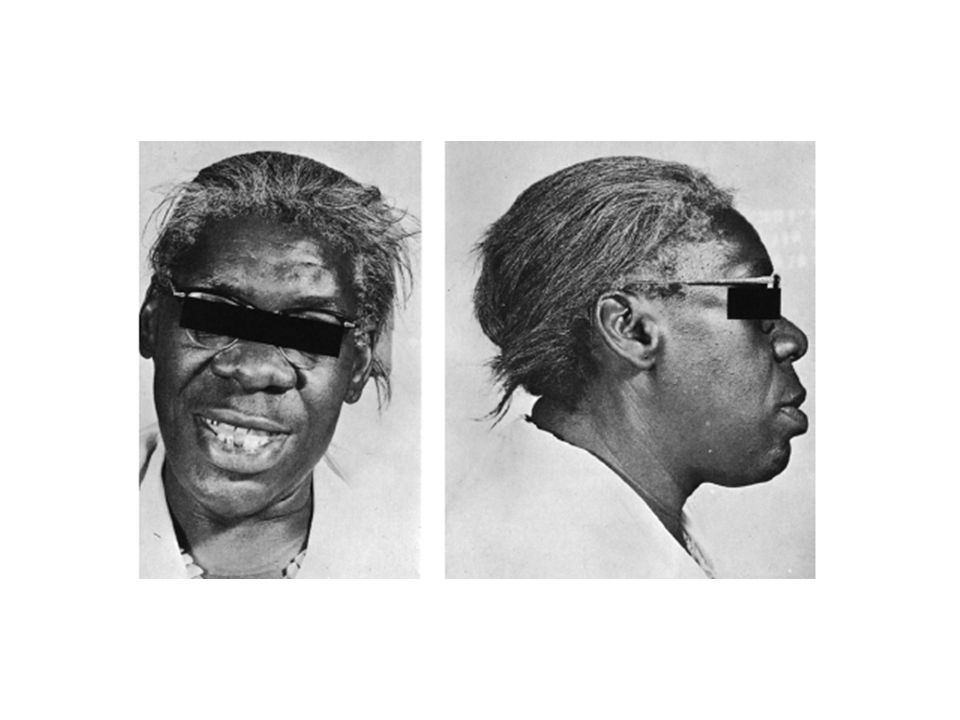
Miastenia gravis Fatigabilidad anormal de la musculatura estriada
Afección inicial de la musculatura externa del ojo Ptosis palpebral uni o bilateral asociada a esfuerzo Diplopía intermitente. Alteraciones de la deglución y la masticación, disfonía y alteraciones del habla. Posteriormente debilidad muscular de las extremidades (proximal). Insuficiencia respiratoria Prueba de Tensilón® (Cloruro de Edrofonio) Anticuerpos contra el receptor de ACh (RIA)
. Insuficiencia respiratoria. Prueba de Tensilón® (Cloruro de Edrofonio) Anticuerpos contra el receptor de ACh (RIA)")
80
Fatiga (Ptosis) en un paciente con MG
mio = músculo astenia = debilidad gravis = intensa.

81
Prueba de Tensilón® (Cloruro de Edrofonio)
")
82
Miastenia gravis Tratamiento Inhibidores de la colinesterasa
Fisostigmina Inmunosupresores Azatioprina Glucocorticoides (Corticoesteroides) Plasmaféresis Timectomía
 Plasmaféresis. Timectomía.")
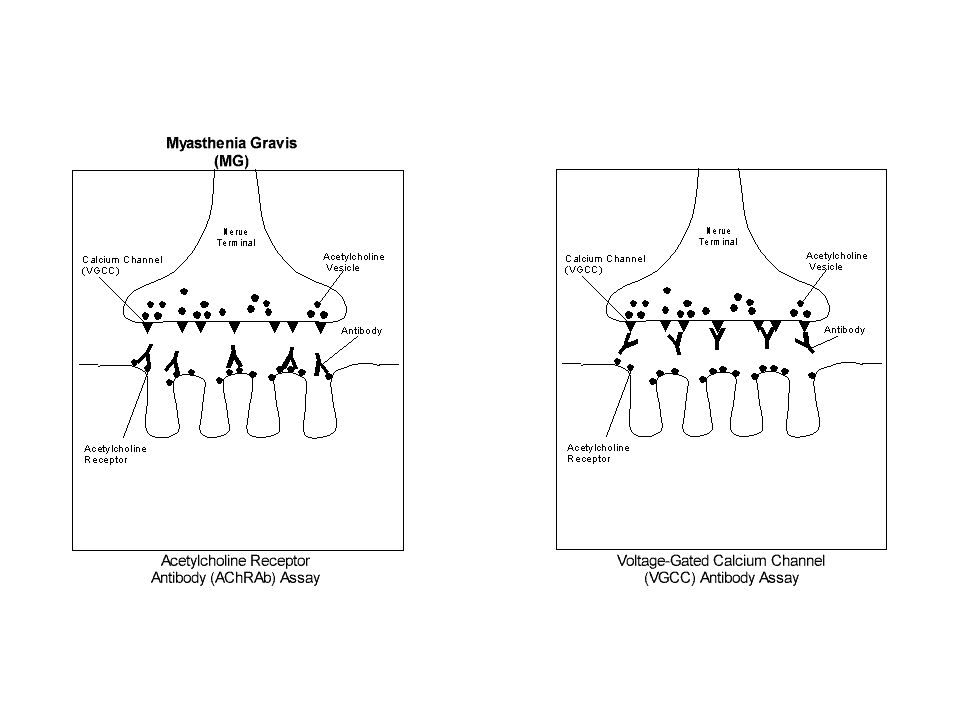
83
Eaton-Lambert. Trastorno adquirido autoinmune
Amplitud del potencial de acción reducido Con estimulación repetitiva se incrementa, llegando a ser varias veces superior al original. Autoanticuerpos frente a los canales de calcio dependientes del voltaje, del nervio motor terminal.

87
Eaton-Lambert. En los varones y en edades superiores a los 40 años,
70% con carcinoma pulmonar del tipo de células pequeñas. Este tumor expresa canales de calcio dependientes del voltaje La sensibilización a estos antígenos conduce a la formación de los anticuerpos que reaccionan de manera cruzada con los canales de calcio de la región neuromuscular presináptica. En mujeres Idiopático Asociado a enfermedad inflamatoria intestinal crónica o granulomatosa.

88
Eaton-Lambert. Debilidad y fatigabilidad proximal
Sobre todo de las extremidades inferiores. Puede existir ptosis palpebral No hay afección ocular Si ocurre, es leve y transitoria. En el 80% de los casos existe disfunción autónoma Disminución de salivación Lagrimeo y sudación Ortostatismo Impotencia Trastornos pupilares

89
Eaton-Lambert. Exploración física comprueba fatigabilidad muscular al esfuerzo repetido. Puede haber ausencia de ROTs Algunos enfermos refieren parestesias. Puede preceder al diagnóstico de neoplasia en años En ocasiones no se modifica con el tratamiento quirúrgico o quimioterápico del tumor. EMG Falta de contracción muscular como respuesta a un estímulo único, o a estímulos de baja frecuencia.

92
Miopatías endocrinas Muchos trastornos endocrinológicos producen debilidad Se relacionan directamente con la alteración hormonal correspondiente Etiología no clara Patogenia en relación con exceso o déficit de la hormona correspondiente Manifestaciones agudas Miopatía crónica

93
Miopatías endocrinas Debilidad muscular rápidamente generalizada
Crisis tirotóxica (Hipertiroidismo) Hipercalcemia (Hiperparatiroidismo) Hipoglicemia (Diabetes Mellitus) Debilidad y atrofia musculares de predominio proximal Hipertiroidismo Síndrome de Cushing
 Hipercalcemia (Hiperparatiroidismo) Hipoglicemia (Diabetes Mellitus) Debilidad y atrofia musculares de predominio proximal. Hipertiroidismo. Síndrome de Cushing.")
94
Miopatías endocrinas Hábito musculoso con escasa fuerza Acromegalia
Hipotiroidismo Fase de relajación prolongada Síndrome de Hoffman Debilidad muscular generalizada con un curso episódico Enfermedad de Addison Síndrome de Conn

95
Miopatías endocrinas Hipotiroidismo Debilidad muscular proximal (1/3)
Contracción y relajación muscular lenta (25%) Fase de relajación prolongada en los reflejos osteotendinosos (ROT) CPK elevada Hiperparatiroidismo Debilidad muscular y atrofia (rara vez se quejan) ROT conservados y vivos
 Fase de relajación prolongada en los reflejos osteotendinosos (ROT) CPK elevada. Hiperparatiroidismo. Debilidad muscular y atrofia (rara vez se quejan) ROT conservados y vivos.")
96
Miopatías endocrinas Hiperparatiroidismo
Debilidad muscular proximal, atrofia y ROT vivos Síndrome de Cushing Debilidad muscular proximal Aspecto cushingoide Acromegalia Debilidad muscular proximal sin atrofia Músculos aumentados de tamaño con menos fuerza

98
Miopatías tóxicas Agudas Crónicas Polimiositis Fibratos Clofibrato
Gemfibrozil Emetina Alcohol Crónicas Estatinas Cloroquina Glucocorticoides Zidovudina (AZT) Polimiositis D-penicilamina
 Polimiositis. D-penicilamina.")
99
Miopatías tóxicas Agudas Crónicas
Debilidad muscular de rápida instalación Calambres Elevación de la CPK Biopsia muestra degeneración muscular reciente Crónicas Debilidad muscular de curso progresivo y lento Atrofia muscular indolora CPK normal o a lo sumo levemente elevada

100
Miopatía alcohólica aguda
Inicio brusco de los síntoma Rápida progresión de éstos en un plazo de 2 semanas. Relacionado con ingestas de grandes cantidades de alcohol. Pueden desarrollarse Rabdomiólisis Mioglobinuria Insuficiencia renal aguda

101
Miopatía por zidovudina (AZT)
Debilidad muscular proximal Con mialgias y moderada o nula elevación de la CK sérica. Relación con la dosis total acumulada (200 g) Fibras deshilachadas de color rojo en porcentajes (fibras AZT) Coexistir con otras miopatías asociadas con HIV-1 Miopatía nemalínica, microvasculitis o polimiositis,
 Fibras deshilachadas de color rojo en porcentajes (fibras AZT) Coexistir con otras miopatías asociadas con HIV-1. Miopatía nemalínica, microvasculitis o polimiositis,")
103
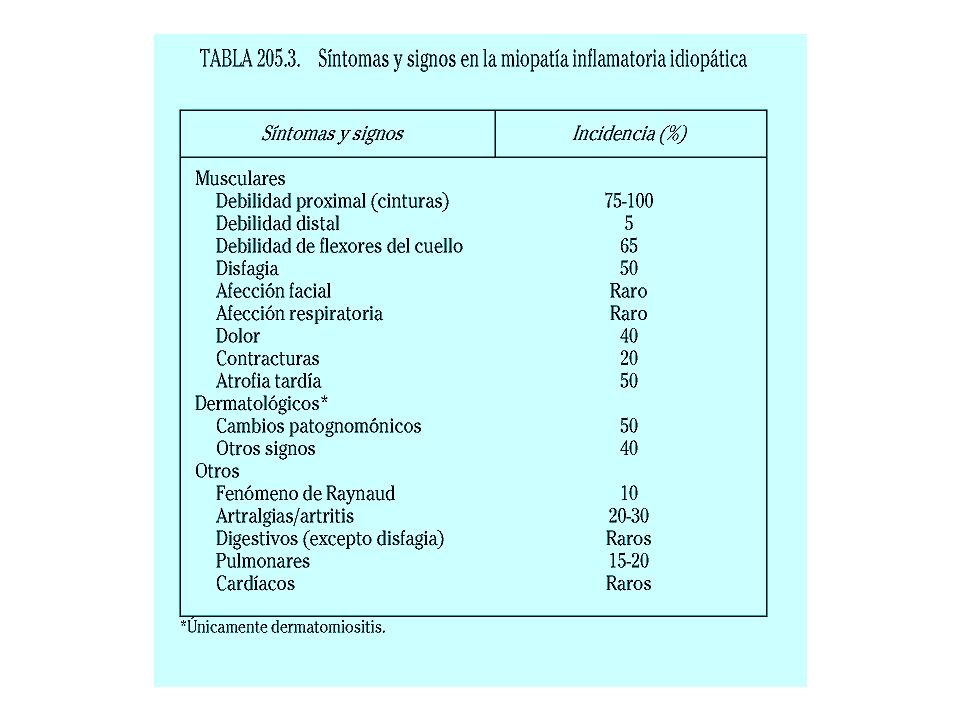
Miopatía inflamatoria Idiopática
Polimiositis/dermatomiositis Polimiositis Dermatomiositis Dermatomiositis asociada con malignidad Dermatomiositis juvenil Asociada a otras enfermedades inmunológicas Lupus eritematoso sistémico (LES) - Esclerodermia (ESP) Miositis por cuerpos de inclusión
 - Esclerodermia (ESP) Miositis por cuerpos de inclusión.")
105
Polimiositis La etiología es desconocida. Patogenia inmunológica
Existen fenómenos de inmunidad celular y humoral Autoanticuerpo circulantes (1/3) responden a los tratamientos inmunodepresores. En la poliomiositis Existencia de fenómenos de citotoxicidad directa Restringida a la expresión por parte del sarcolema de antígenos de clase I del CMH.
 responden a los tratamientos inmunodepresores. En la poliomiositis. Existencia de fenómenos de citotoxicidad directa. Restringida a la expresión por parte del sarcolema de antígenos de clase I del CMH.")
106
Polimiositis En la dermatomiositis
Disminución en el número total de capilares, así como en la densidad capilar Se ha demostrado lesión capilar con depósito de C5b9 (complejo de ataque de membrana). Se detecta precozmente Existe incluso en aquellos casos en los que aún no hay lesión cutánea evidente.
. Se detecta precozmente. Existe incluso en aquellos casos en los que aún no hay lesión cutánea evidente.")
108
Polimiositis Laboratorio Las enzimas musculares muy elevadas
CPK, DHL y aldolasa. ANA + (50-80%) Jo1 (Histidil-tARN sintetasa) Mi-2 PM/scl anti-SRP Electromiografía característica Potenciales de fibrilación y un patrón típicamente "miopático" Biopsia muscular
 Jo1 (Histidil-tARN sintetasa) Mi-2. PM/scl. anti-SRP. Electromiografía característica. Potenciales de fibrilación y un patrón típicamente miopático Biopsia muscular.")
109
Polimiositis Criterios diagnósticos (Bohan y Peter, 1975)
Debilidad proximal, simétrica En las cinturas y los flexores del cuello Semanas o meses de evolución Biopsia muscular Necrosis, fagocitosis, regeneración e infiltrado inflamatorio Puede existir atrofia perifascicular, asimetría en el tamaño de las fibras y alteraciones de tipo oxidativo

110
Polimiositis Elevación de las enzimas musculares
CPK, pero también aldolasa, SGOT y LDH Potenciales de unidad motora polifásicos, fibrilación y "puntas" breves en el EMG. Manifestaciones en piel Coloración violácea de párpados (heliotropo), con edema periorbitario Signo de Gottron Dermatitis eritematosa descamativa en el dorso de las manos (MCF e IF) MCF: metacarpofalángicas IF: interfalángicas
, con edema periorbitario. Signo de Gottron. Dermatitis eritematosa descamativa en el dorso de las manos (MCF e IF) MCF: metacarpofalángicas. IF: interfalángicas.")
111
Dermatomiositis Complejos inmunes
Debilidad muscular proximal y simétrica Manifestaciones en piel: Eritema en heliotropo Pápulas de Gottron Eritema en chal

Presentaciones similares