Descargar la presentación
La descarga está en progreso. Por favor, espere

1
Aspectos Claves de la Evolución Molecular 1. Porqué estudiar filogenética y evolución molecular 2. Aspectos claves: C. genético, Mutaciones. 3. Alineamientos y homología 4. Construcción de árboles filogenéticos 5. Modelos de sustitución Porqué estudiar filogenética y evolución molecular? “Nothing in biology makes sense except in the light of evolution” - Theodosius Dobzhanski, 1973 (The American Biology Teacher 35:125) “Nothing in evolutionary biology makes sense except in the light of a phylogeny” - Jeff Palmer, Douglas Soltis, Mark Chase, 2004 (American J. Botany 91: 1437-1445)
 Nothing in evolutionary biology makes sense except in the light of a phylogeny - Jeff Palmer, Douglas Soltis, Mark Chase, 2004 (American J. Botany 91: ).")
2
The evolutionary thinking Russel Wallace writes to Charles Darwin (June 17 th 1858) u Ernst Haeckel (mid- 19 th Century): the tree of life u The neo-synthesis (Fisher, Heldane, and Wright, 1930-1950)
 u Ernst Haeckel (mid- 19 th Century): the tree of life u The neo-synthesis (Fisher, Heldane, and Wright, )")
3
The molecular RE volution Nuttal, 1904: Serological cross-reactions to study phylogenetic relationships among various group of animals. Watson and Crick beautiful helix! Zuckerland and Pauling, 1965: molecular clocks. Fitch & Margoliash, 1967: Construction of phylogenetic trees.A method based on mutation distances as estimated from cytochrome c sequences is of general applicability (Science, 155:279-284). Kimura, 1968: Evolutionary rate at the molecular level (Nature, 217:624-626). The birth of molecular evolution
. Kimura, 1968: Evolutionary rate at the molecular level (Nature, 217: ). The birth of molecular evolution.")
4
Genetic code In the RNA that encode a protein, each triplet of bases is recognized by the ribosome as a code for a specific amino acid. This genetic code is universal for all organisms, with only a few exceptions such as the mitochondria. There are 64 possible triplets: 61 sense codons (encode 20 amino acids) and 3 non-sense codons (stop codons). A reading frame that is able to encode for a protein (open reading frame, ORF) starts with a codon for methionine and ends with a stop codon. Inferencia Filogenética Aspectos claves: C. genético, Mutaciones.
 and 3 non-sense codons (stop codons). A reading frame that is able to encode for a protein (open reading frame, ORF) starts with a codon for methionine and ends with a stop codon. Inferencia Filogenética Aspectos claves: C. genético, Mutaciones..")
5
Point Mutations Errors in duplication of genetic information can result in the incorporation of a noncomplementary nucleotide: point mutations. Point mutations at the 1 st, 2 nd, and 3 rd codon position usually (96%), always (100%), and rarely (30%) result in an amino-acid change, respectively. Point mutations that do not results in an amino-acid change are called synonymous. Point mutations that results in an amino-acid change are called non-synonymous. Inferencia Filogenética Aspectos claves: C. genético, Mutaciones.
, always (100%), and rarely (30%) result in an amino-acid change, respectively. Point mutations that do not results in an amino-acid change are called synonymous. Point mutations that results in an amino-acid change are called non-synonymous. Inferencia Filogenética Aspectos claves: C. genético, Mutaciones..")
6
A Transitions and transversions C TG u Transitions ( ) are purine (A, G) or pyrimidine (C, T) mutations: Pu- Pu, Py-Py u Transversions ( ) are purine to pyrimidine mutations or the reverse: (Pu-Py, or Py-Pu).
 are purine (A, G) or pyrimidine (C, T) mutations: Pu- Pu, Py-Py u Transversions ( ) are purine to pyrimidine mutations or the reverse: (Pu-Py, or Py-Pu).")
7
u 4 possible transitions: A G, C T u 8 possible transversions: A C, A T, G C, G T u Thus if mutations were random, transversions are 2 times more likely than transitions. u Due to steric hindrance (as well as negative selection!), the opposite is true, transitions occur in general more often than transversions [2-15 times more, depending on the gene region and the species]. Point mutations and the genetic code
, the opposite is true, transitions occur in general more often than transversions [2-15 times more, depending on the gene region and the species]. Point mutations and the genetic code.")
8
Indel Mutations Errors in duplication of genetic information can also result in deletions or insertions of one or more nucleotides: indels mutations. When three (or multiples of three) nucleotides are inserted or deleted in coding regions, the ORF remains intact, but one (or more) amino acids are inserted or deleted. In any other case, indels mutations disturb the ORF and the resulting gene codes for an entirely different protein, with a different length than the original one. Viruses often encode several proteins from a single gene by using overlapping ORFs. Inferencia Filogenética Aspectos claves: C. genético, Mutaciones.
 nucleotides are inserted or deleted in coding regions, the ORF remains intact, but one (or more) amino acids are inserted or deleted. In any other case, indels mutations disturb the ORF and the resulting gene codes for an entirely different protein, with a different length than the original one. Viruses often encode several proteins from a single gene by using overlapping ORFs. Inferencia Filogenética Aspectos claves: C. genético, Mutaciones..")
10
Mecanismos genéticos explotados por los virus para la generación de variabilidad

11
Mutacion e Hipermutación Inserciones y Delecciónes Recombinación Reordenamientos

12
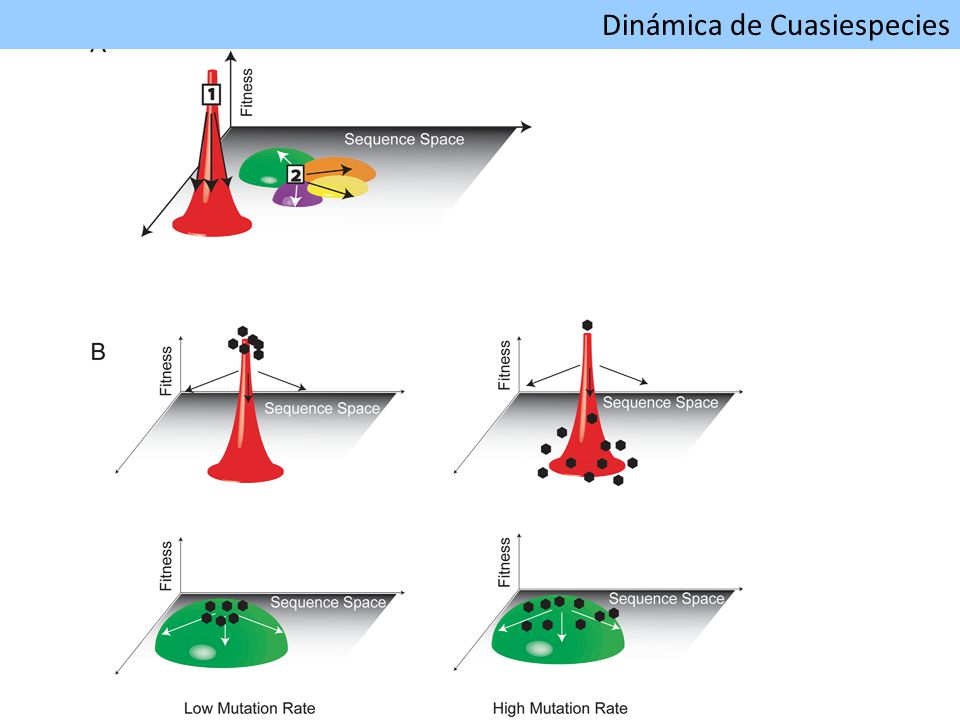
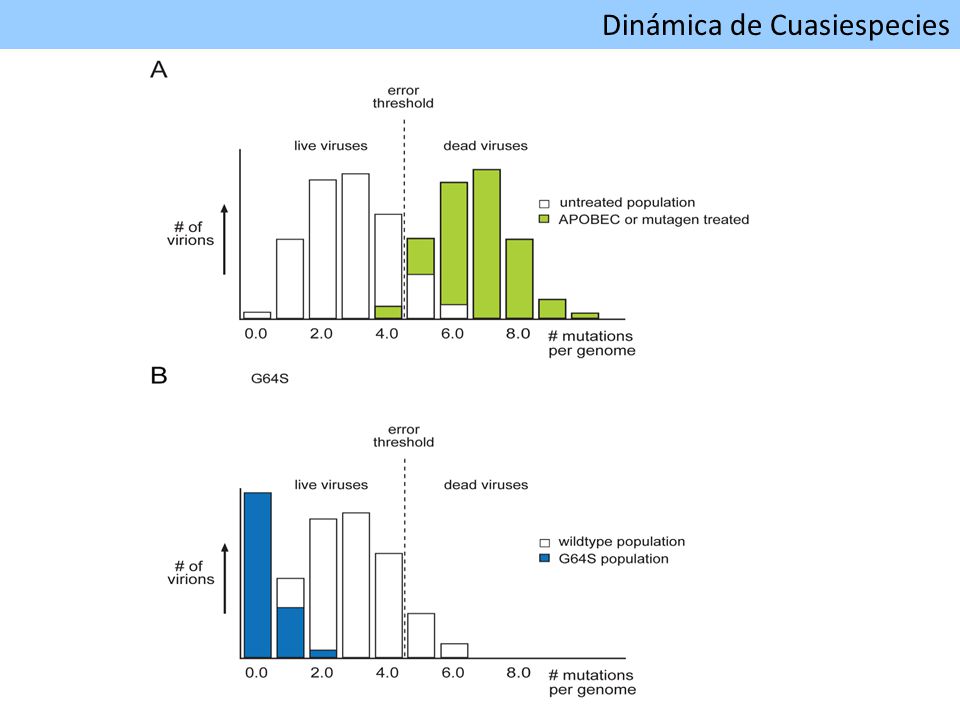
Dinámica de Cuasiespecies

13
compleja población de variantes fuertemente relacionadas genéticamente baja fidelidad de la ARN polimerasa ARN dependiente Secuencia maestra Concentración Espacio Secuencia Espectro de mutantes Schuster P 2008 Dinámica de Cuasiespecies

16
Cooperación y complementación Dinámica de Cuasiespecies

19
Alignments There are three main methods of sequence alignments: 1) Manual 2) Automatic (Dynamic programming, Progressive alignment) 3) Combined Inferencia Filogenética Alineamientos y homología
 Manual 2) Automatic (Dynamic programming, Progressive alignment) 3) Combined Inferencia Filogenética Alineamientos y homología")
20
Alignments An alignment is an hypothesis of positional homology between nucleotide/amino acids. Homologous sequences are usually aligned such that homologous sites form columns in the alignment. Easy Difficult due to indels Human beta PEEKSAVTALWGKV Horse beta GEEKAAVLALWDKV Human alpha PADKTNVKAAWGKV Horse alpha AADKTNVKAAWSKV Whale myoglobin EGEWQLVLHVWAKV Lamprey globin AAEKTKIRSAWAPV Lupin globin ESQAALVKSSWEEF Human beta VHLTN–-FFESFGDLST Horse beta VQLSN–-FFDSFGDLSN Human alpha -VLSGAHYFPHF-DLS- Horse alpha -VLSGGHYFPHF-DLS- Whale myoglobin -VLSEADKFDRFKHLKT Lamprey globin APLSYSTFFPKFKGLTT Lupin globin GALTNANLFSFLKGTSE Inferencia Filogenética Alineamientos y homología

21
Dynamic programming Dynamic programming (Needleman and Wunsch, 1970; Gotoh, 1982) is an exhaustive method that find the best alignment by giving substitutions scores for all pairs of aligned residues and gap penalties (GP). To prevent excessive use of gaps, indels are usually penalized using so-called GP. Alignment programs have separate penalties for inserting a gap (gap opening) and for extending a gap (gap extend). G A T T T CG A T – T T C G A A T T CG A – A T T C Inferencia Filogenética Alineamientos y homología
 and for extending a gap (gap extend). G A T T T CG A T – T T C G A A T T CG A – A T T C Inferencia Filogenética Alineamientos y homología.")
22
Scoring Scheme: Match: +1 Mismatch: 0 Indel: -1 Dynamic programming Inferencia Filogenética Alineamientos y homología

23
G A T T C – G A A T T C 1 1 0 1 0 Alignment score = 2 Dynamic programming Alineamientos y homología

24
G A – T T C G A A T T C Alignment score = 4 Dynamic programming 1 1 1 1 1 Alineamientos y homología

25
G – A T T C G A A T T C Alignment score = 4 Dynamic programming 1 1 1 1 1 Alineamientos y homología

26
Multiple Sequence Alignments Phylogenetic trees are based on multiple sequence alignments. Dynamic programming can be used to align multiple sequences but the time required growth exponentially with the number of sequences. Until end 1989 multiple sequences alignments were assembled by hand because the exhaustive alignment of more than five or six sequences is computationally unfeasible. Now, most multiple sequences alignments are constructed by the method known as progressive sequence alignment (Feng and Doolittle, 1987; Higgins and Sharp; 1988). Alineamientos y homología
. Alineamientos y homología.")
27
Progressive Alignment Progressive alignment is a heuristic method as it makes no guarantees to produce an alignment with the best score according to a formula. 1) Perform all possible pairwise alignments between each pair of sequences using a fast/approximate method. 2) Calculate the ‘distance’ between each pair of sequences and construct a crude “guide tree” with the Neighbor-Joining method. 3) The alignment is gradually built up by following the branching order in the tree, with each step being treated as a dynamic programming pairwise alignment, sometimes with each member of a ‘pair’ having more than one sequence. Alineamientos y homología
 Perform all possible pairwise alignments between each pair of sequences using a fast/approximate method. 2) Calculate the ‘distance’ between each pair of sequences and construct a crude guide tree with the Neighbor-Joining method. 3) The alignment is gradually built up by following the branching order in the tree, with each step being treated as a dynamic programming pairwise alignment, sometimes with each member of a ‘pair’ having more than one sequence. Alineamientos y homología.")
28
Progressive alignment - step 1 1. gctcgatacgatacgatgactagcta 2. gctcgatacaagacgatgacagcta 3. gctcgatacacgatgacta----gcta 4. gctcgatacacgatgacga---gcga 5. ctcgaacgatacgatgact----agct 1. gctcgatacgatacgatgactagcta 2. gctcgatacaagacgatgac-agcta 1 2 3 4 5 Alineamientos y homología

29
1. gctcgatacgatacgatgactagcta 2. gctcgatacaagacgatgac-agcta 3. gctcgatacacgatgactagcta 4. gctcgatacacgatgacgagcga 5. ctcgaacgatacgatgactagct 3. gctcgatacacgatgactagcta 4. gctcgatacacgatgacgagcga 1 2 3 4 5 Progressive alignment - step 2 Alineamientos y homología

30
1. gctcgatacgatacgatgactagcta 2. gctcgatacaagacgatgac-agcta + 3. gctcgatacacgatgactagcta 4. gctcgatacacgatgacgagcga 1. gctcgatacgatacgatgactagcta 2. gctcgatacaagacgatgac-agcta 3. gctcgatacacga---tgactagcta 4. gctcgatacacga---tgacgagcga 1 2 3 4 5 Progressive alignment - step 3 Alineamientos y homología

31
1. gctcgatacgatacgatgactagcta 2. gctcgatacaagacgatgac-agcta 3. gctcgatacacga---tgactagcta 4. gctcgatacacga---tgacgagcga + 5. ctcgaacgatacgatgactagct 1. gctcgatacgatacgatgactagcta 2. gctcgatacaagacgatgac-agcta 3. gctcgatacacga---tgactagcta 4. gctcgatacacga---tgacgagcga 5. -ctcga-acgatacgatgactagct- 1 2 3 4 5 Progressive alignment – final step Alineamientos y homología

32
. Construcción de árboles filogenéticos La inferencia de relaciones filogenéticas a partir de secs. moleculares requiere de la selección de uno de los muchos métodos disponibles Con frecuencia la inferencia filogenética es considerada como una “caja negra” en la que “entran las secuencias y salen los árboles” La inferencia de relaciones filogenéticas a partir de secs. moleculares requiere de la selección de uno de los muchos métodos disponibles Con frecuencia la inferencia filogenética es considerada como una “caja negra” en la que “entran las secuencias y salen los árboles” Objetivos fundamentales 1. desarrollar un marco conceptual para entender los fundamentos teóricos (filosóficos) que distinguen a los distintos métodos de inferencia (clasificación de métodos) 2. presentar el uso de modelos y suposiciones en filogenética
 que distinguen a los distintos métodos de inferencia (clasificación de métodos) 2. presentar el uso de modelos y suposiciones en filogenética.")
34
Tree Reconstructions Methods Maximum-likelihood Bayesian Inference Neighbour-Joining Minimum Evolution UPGMA Maximum-parsimony Character-based methodsDistance-based methods Methods based on an explicit model of evolution Methods not based on an explicit model of evolution. Construcción de árboles filogenéticos

35
Neighbor-Joining method The NJ (Saitou and Nei, 1987) method is a heuristic method for estimating the minimum evolution tree. The NJ method is based on the minimum evolution principle and construct internal nodes by joining nearest neighbors (two taxa connected by a single node) in each step. Distance Matrix. Construcción de árboles filogenéticos
 in each step. Distance Matrix. Construcción de árboles filogenéticos.")
36
Distance 3.3 (Human - Monkey) is the minimum. So we'll join Human and Monkey to MonHum. Mon-Hum MonkeyHumanSpinachMosquitoRice Neighbor-Joining (1) Recalculate the distance matrix again…... Construcción de árboles filogenéticos
 Recalculate the distance matrix again…... Construcción de árboles filogenéticos.")
37
HumanMosquito Mon-Hum MonkeySpinachRice Mos-(Mon-Hum) Neighbor-Joining (2) Recalculate the distance matrix again…... Construcción de árboles filogenéticos

38
HumanMosquito Mon-Hum MonkeySpinachRice Mos-(Mon-Hum) Spin-Rice Neighbor-Joining (3) Recalculate the distance matrix again…... Construcción de árboles filogenéticos

39
HumanMosquito Mon-Hum MonkeySpinachRice Mos-(Mon-Hum) Spin-Rice (Spin-Rice)-(Mos-(Mon-Hum)) Neighbor-Joining (4). Construcción de árboles filogenéticos

40
Human Monkey Mosquito Rice Spinach Unrooted Neighbor-Joining Tree. Construcción de árboles filogenéticos

41
Distance-based methods Advantages: - very fast - allows the use of an explicit model of evolution Disadvantages: - only produces one best tree (we do not get any idea about other potential tress) - reduces all sequence information into a single distance value - generally outperformed by Maximum likelihood or Bayesian methods in choosing the correct tree in computer simulations. Construcción de árboles filogenéticos

42
Tree Reconstructions Methods Maximum-likelihood Bayesian Inference Neighbour-Joining Minimum Evolution UPGMA Maximum-parsimony Character-based methodsDistance-based methods Methods based on an explicit model of evolution Methods not based on an explicit model of evolution. Construcción de árboles filogenéticos Neighbour-Joining

43
. Construcción de árboles filogenéticos

Presentaciones similares




 Hacer: to make/do.>")
>")




. When we ask what time it is in Spanish, we say “¿Qué hora es?” Some people also say “¿Qué horas son?”>")












