Descargar la presentación
La descarga está en progreso. Por favor, espere

1
LECCIÓN 14: INTRODUCCIÓN A LA ESPECTROSCOPÍA MOLECULAR
La radiación electromagnética y su interacción con la materia. Regiones del espectro y tipos de espectroscopías. Absorción y emisión de radiación. La ley de Lambert-Beer y anchura de banda espectral. Espectroscopía de rotación. Espectroscopía de vibración. Espectroscopía electrónica. Espectroscopía fotoelectrónica (UPS). Otros tipos de espectroscopía.
. Otros tipos de espectroscopía.")
2
Incidencia sobre un medio no absorbente (sin considerar la reflexión)
Propagación de la luz Incidencia sobre un medio no absorbente (sin considerar la reflexión) Medio no absorbente. n – índice de refracción s – velocidad de la luz en el medio Incidencia sobre un medio absorbente Medio absorbente. N – índice de refracción complejo α – Coeficiente de absorción Abs – Absorbancia d – espesor del film absorbente K – cte de absorción
 Medio no absorbente. n – índice de refracción. s – velocidad de la luz en el medio. Incidencia sobre un medio absorbente. Medio absorbente. N – índice de refracción complejo. α – Coeficiente de absorción. Abs – Absorbancia. d – espesor del film absorbente. K – cte de absorción.")
3
La radiación electromagnética y su interacción con la materia.
-Reflexión: En un sólido especular: -Dispersión< -Transmisión: incidencia ≠ 90º, refractancia. -Absorción: Espectroscopia: Todos los fenómenos de absorción y emisión de radiación por parte de la materia. Espectroscopias de absorción: En la medida de IA, se basan las espectroscopias de absorción (microondas, IR y electrónica), aunque existen otras técnicas que se basan en las medidas de IR. En la medida de ID =Rayos X, Scatering (dispersión de partículas) Se suelen denominar espectroscopia, aunque no existe absorción como fenómeno fundamental Espectroscopias de emisión: Las moléculas son previamente excitadas, analizándose la radiación que emiten al retornar a su estado fundamental. La excitación puede ser: -Por una reacción química (quimiluminiscencia) -Por absorción de radiación (fluorescencia y fosforescencia). - Mediante el paso de una corriente eléctrica (descarga voltaica en un medio enrarecido). -Aplicación de campos magnéticos (técnicas de resonancia). Espectroscopia Fotoelectrónica (ionización)
, aunque existen otras técnicas que se basan en las medidas de IR. En la medida de ID =Rayos X, Scatering (dispersión de partículas) Se suelen denominar espectroscopia, aunque no existe absorción como fenómeno fundamental. Espectroscopias de emisión: Las moléculas son previamente excitadas, analizándose la radiación que emiten al retornar a su estado fundamental. La excitación puede ser: -Por una reacción química (quimiluminiscencia) -Por absorción de radiación (fluorescencia y fosforescencia). - Mediante el paso de una corriente eléctrica (descarga voltaica en un medio enrarecido). -Aplicación de campos magnéticos (técnicas de resonancia). Espectroscopia Fotoelectrónica (ionización)")
4
Regiones del espectro y tipos de espectroscopías.
kBT En el UV-visible, es común caracterizar el espectro mediante la longitud de onda en nanometros (1nm=10-9m). El visible abarca desde 700 a 400 nm. En el IR, se suele utilizar el micrometro (1 m = m), o número de ondas , en cm-1. Así, para = 1013 s-1, → = 333 cm-1. Un espectro típico abarca desde los 400 a 4000 cm-1.
. El visible abarca desde 700 a 400 nm. En el IR, se suele utilizar el micrometro (1 m = 10-6 m), o número de ondas , en cm-1. Así, para = 1013 s-1, → = 333 cm-1. Un espectro típico abarca desde los 400 a 4000 cm-1.")
5
Energía acumulada promedio = kBT
Reglas de selección Energía acumulada promedio = kBT La intensidad de una banda depende de la población del nivel ley de distribución de Maxwell-Boltzmann: Si En << kBT → Nn > 0 Si En >> kBT → Nn = 0 Para T = 25ºC, kBT = Julios/molécula. Esta energía es equivalente a = kBT/h = s-1, frecuencia que corresponde al IR lejano

6
Absorción y emisión de radiación.
Absorción inducida de radiación: El campo eléctrico del fotón (E ) interacciona con el momento dipolar de la molécula ( µ) perturbando el Hamiltoniano del sistema. Según la teoría de perturbaciones µnm es el dipolo de transición. La probabilidad del salto espectroscópico viene dada por el coeficiente de absorción inducida de Einstein:
 interacciona con el momento dipolar de la molécula ( µ) perturbando el Hamiltoniano del sistema. Según la teoría de perturbaciones. µnm es el dipolo de transición. La probabilidad del salto espectroscópico viene dada. por el coeficiente de absorción inducida de Einstein:")
7
La emisión de radiación puede tener lugar mediante dos mecanismos diferentes, de forma espontánea, o de forma inducida Estado estacionario la fuente de la radiación incidente es térmica ley de distribución de Maxwell-Boltzmann

8
Para < 1012s-1 puede despreciarse la emisión espontánea
Si la frecuencia es alta (UV-visible e IR cercano), domina la emisión espontánea. Si la frecuencia es baja (IR lejano, microondas, radioondas etc), la emisión es fundamentalmente inducida. La emisión espontánea dirección al azar Emisión inducida en fase y misma dirección que la incidente Para < 1012s-1 puede despreciarse la emisión espontánea Emisión espontanea Emisión inducida Las técnicas de resonancia (RMN y RSE), y el efecto Laser, están basados en las propiedades de la emisión inducida. Cuando nm es pequeño, las moléculas pueden permanecer en estados excitados durante periodos de tiempo indefinidos, ya que la probabilidad de emisión espontánea es despreciable.
, domina la emisión espontánea. Si la frecuencia es baja (IR lejano, microondas, radioondas etc), la emisión es fundamentalmente inducida. La emisión espontánea dirección al azar. Emisión inducida en fase y misma dirección que la incidente. Para < 1012s-1 puede despreciarse la emisión espontánea. Emisión espontanea. Emisión inducida. Las técnicas de resonancia (RMN y RSE), y el efecto Laser, están basados en las propiedades de la emisión inducida. Cuando nm es pequeño, las moléculas pueden permanecer en estados excitados durante periodos de tiempo indefinidos, ya que la probabilidad de emisión espontánea es despreciable.")
9
La muestra es sometida a la acción de un campo magnético B0, el cual orienta los spines nucleares en dos direcciones, o a favor, o en contra, casi al 50%. Las moléculas excitado no emiten radiación de forma espontánea. Para que la emisión se produzca, debe ser inducida Para inducir la emisión, se sitúa una bobina a lo largo del eje x (90º con respecto a la dirección del campo magnético B0) y se hace pasar por ella una corriente alterna de frecuencia variable, =2, próxima a 0=20.
 y se hace pasar por ella una corriente alterna de frecuencia variable, =2, próxima a 0=20.")
10
La ley de Lambert-Beer y anchura de banda espectral.
F0 F+dF F Ley de Lambert-Beer dx En medios homogéneos, A es proporcional a la concentración c, y a la longitud del paso óptico. es una constante de proporcionalidad denominada absortibidad molar, que es función de la frecuencia

11
En los espectros aparecen bandas de anchura muy variable
- Transiciones simultaneas electrónicas, vibracionales y rotacionales -Efecto Doppler en sistemas gaseosos. - Indeterminación en la energía de los estados ∆E, es inversamente proporcional a la incertidumbre en el tiempo de vida media de dicho estado. Si el tiempo de vida media de un estado es infinito (estado estable) su energía puede conocerse exactamente. Si t→ 0, la indeterminación en la energía es grande (anchura de banda espectral)
 su energía puede conocerse exactamente. Si t→ 0, la indeterminación en la energía es grande (anchura de banda espectral)")
12
Espectroscopía de rotación.
En la Lección 5, se vio que la energía de rotación de una molécula diatómica era: Espectroscopía de rotación. Las funciones de onda eran los armónicos esféricos (parte angular del átomo de hidrógeno) que dependía de m y ℓ. Sin embargo la energía depende solo de ℓ Para no confundir ℓ y m con los nºs cuánticos atómicos, en rotación se les llama J y M. Cada uno de los estados J, está 2J+1 veces degenerado. Las reglas de selección en rotación son ΔJ = ±1. La molécula debe tener momento dipolar permanente. Moléculas como O2 y N2 no son activas. Saltos permitidos para la absorción J+1 J
 que dependía de m y ℓ. Sin embargo la energía depende solo de ℓ. Para no confundir ℓ y m con los nºs cuánticos atómicos, en rotación se les llama J y M. Cada uno de los estados J, está 2J+1 veces degenerado. Las reglas de selección en rotación son ΔJ = ±1. La molécula debe tener momento dipolar permanente. Moléculas como O2 y N2 no son activas. Saltos permitidos para la absorción. J+1. J.")
13
El nivel más poblado → d[NJ/N]/dJ = 0
Si I aumenta B disminuye. En molécula de alto Pm B → 0 2B La técnica permite determinar distancias y ángulos de enlace en moléculas pequeñas La altura de las bandas depende de la población del nivel de partida. Distribución de Maxweell-Boltzmann El nivel más poblado → d[NJ/N]/dJ = 0
![El nivel más poblado → d[NJ/N]/dJ = 0](http://slideplayer.es/slide/1022609/2/images/13/El+nivel+m%C3%A1s+poblado+%E2%86%92+d%5BNJ%2FN%5D%2FdJ+%3D+0.jpg "Si I aumenta B disminuye. En molécula de alto Pm B → 0. 2B. La técnica permite determinar distancias y ángulos de enlace en moléculas pequeñas. La altura de las bandas depende de la población del nivel de partida. Distribución de Maxweell-Boltzmann. El nivel más poblado → d[NJ/N]/dJ = 0.")
14
La energía de rotación de una molécula poliatómica no lineales es:
Pueden obtenerse los tres momentos de inercia de una molécula en estado gaseoso. Cuando la molécula posee pocos átomos, es posible calcular distancias interatómicas y/o ángulos de enlace. En moléculas complejas dicho tipo de cálculo no es posible, ya que solo se dispone de tres datos experimentales (momentos de inercia). En función de su geometría, las moléculas pueden clasificarse como Lineales: Iz =0 y Iy = Ix Trompo esféricas: Iz = Iy = Ix, (CH4). Sin momento dipolar permanente, no son activas en microondas. Trompo simétricas: Alargadas, Iz < Iy = Ix, como el CH3Cl. Achatadas, Iz = Iy < Ix, como BF3 o CHCl3 Trompo asimétricas, Iz < Iy < Ix. La mayor parte de las moléculas pertenecen a este grupo Cuando la molécula tiene elementos de simetría existen relaciones entre los diferentes momentos de inercia. Así, en el agua Ix = Iy + Iz.
. En función de su geometría, las moléculas pueden clasificarse como. Lineales: Iz =0 y Iy = Ix. Trompo esféricas: Iz = Iy = Ix, (CH4). Sin momento dipolar permanente, no son activas en microondas. Trompo simétricas: Alargadas, Iz < Iy = Ix, como el CH3Cl. Achatadas, Iz = Iy < Ix, como BF3 o CHCl3. Trompo asimétricas, Iz < Iy < Ix. La mayor parte de las moléculas pertenecen a este grupo. Cuando la molécula tiene elementos de simetría existen relaciones entre los diferentes momentos de inercia. Así, en el agua Ix = Iy + Iz.")
15
-Solo existen moléculas pequeñas
-T muy baja (J bajo) -Las moléculas se detectan por su I Estrella Gas interestelar
 -Las moléculas se detectan por su I. Estrella. Gas interestelar.")
16
Espectroscopía de vibración.
Moléculas diatómicas. Modelo del oscilador armónico υ, frecuencia característica de cada enlace (IR) v = 0, 1, 2,.. Separación entre dos niveles consecutivos, Ev+1 -Ev = hυ, es constante. hυ > kBT, a temperatura ambiente, Las moléculas se sitúan en el nivel fundamental, v = 0. Las moléculas pueden situarse en estados excitados de vibración mediante choques con otras moléculas, o mediante absorción de radiación IR
 v = 0, 1, 2,.. Separación entre dos niveles consecutivos, Ev+1 -Ev = hυ, es constante. hυ > kBT, a temperatura ambiente, Las moléculas se sitúan en el nivel fundamental, v = 0. Las moléculas pueden situarse en estados excitados de vibración mediante choques con otras moléculas, o mediante absorción de radiación IR.")
17
La regla de selección es Δv = ±1 (modelo del oscilador armónico)
Una molécula diatómica debe poseer momento dipolar permanente para que pueda absorber radiación IR. Por tanto, moléculas como H2, O2 y N2 son transparentes a esta radiación. Como las moléculas están en v = 0, el único salto permitido será desde v=0 a v=1. Sin embargo las moléculas no son auténticos asciladores armónicos. Pueden darse (sobretonos o armónicos) donde Δv > 1 La probabilidad disminuye conforme Δv aumenta. Con fines prácticos, una molécula diatómica solo posee una banda en espectroscopía IR, la que corresponde al salto desde v=0 a v=1.
 donde Δv > 1. La probabilidad disminuye conforme Δv aumenta. Con fines prácticos, una molécula diatómica solo posee una banda en espectroscopía IR, la que corresponde al salto desde v=0 a v=1.")
18
Espectro del CO2 en fase gas

19
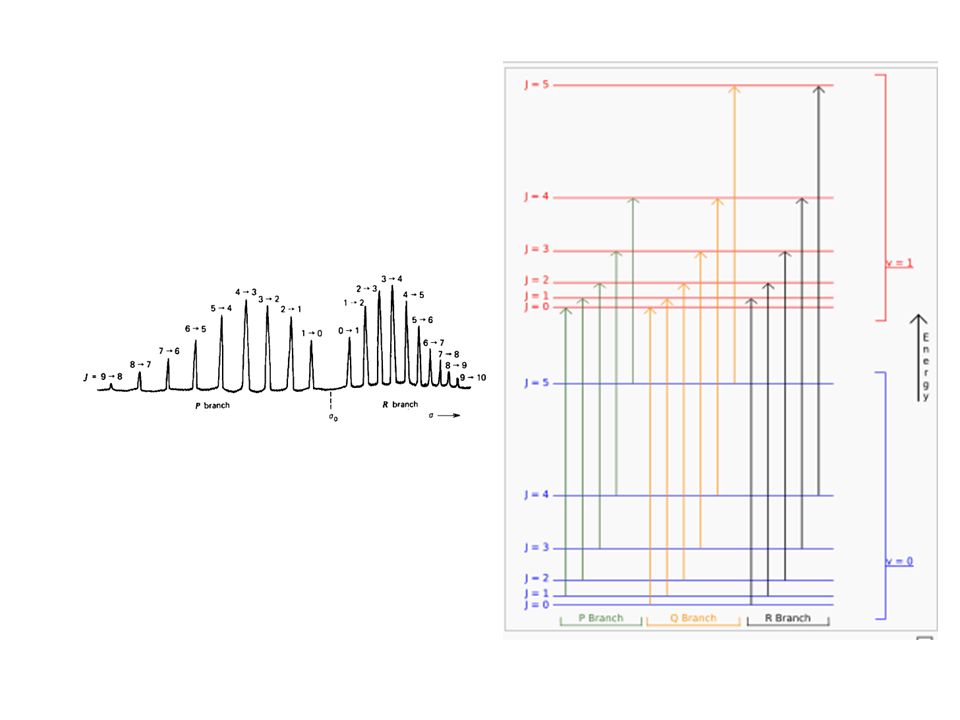
Espectro IR de transmisión del CO gaseoso
El espectro posee una forma típica, que consta de una gran cantidad de pequeñas líneas divididas en dos grandes grupos. Espectro IR de Absorción del ClH gaseoso La separación entre niveles de energía de rotación es muy inferior a la de los niveles de energía de vibración, por ello, cuando una molécula absorbe un fotón para pasar de v=0 a v=1 simultáneamente, se produce el salto entre niveles de rotación ΔJ = ±1

21
A medida que la molécula es mayor, sus momentos de inercia también lo son, y la constante B es cada vez más pequeña, por lo que la separación entre las líneas rotacionales no puede distinguirse, observándose solo la silueta de la banda.

22
Las moléculas con más de dos atómicas pueden vibrar de más de un modo.
Para describir el movimiento de una molécula de N átomos, se necesitan 3N coordenadas. Moléculas lineales: 3 son de traslación 2 de rotación 3N-5 modos de vibración Moléculas no lineales: 3 son de traslación 3 de rotación 3N-6 modos de vibración Tensión simétrica (Inactiva en IR) Tensión simétrica Tensión asimétrica Tensión asimétrica Cada uno de estos modos vibra a una frecuencia diferente, y da lugar en espectroscopía IR a una banda diferente.
 Tensión simétrica. Tensión asimétrica. Tensión asimétrica. Cada uno de estos modos vibra a una frecuencia diferente, y da lugar en espectroscopía IR a una banda diferente.")
23
La tensión asimétrica pertenece al grupo b2.
En los modos normales de vibración todos los átomos vibran en fase y con la misma frecuencia. Las vibraciones reales de las moléculas son muy complejas, si bien una molécula solo absorbe radiación para modificar uno de sus modos normales, es decir, cuando la vibración de los átomos está en fase. Tensión simétrica Tensión asimétrica Los modos de vibración de una molécula suelen denominarse en base a la teoría de grupos, En el caso del agua, la tensión simétrica y la flexión simétrica tienen simetría a1 (permanecen invariantes al rotar con respecto a C2 y al efectuar la reflexión σv1), La tensión asimétrica pertenece al grupo b2.
, La tensión asimétrica pertenece al grupo b2.")
24
Los espectros reales son más complejos: -sobretonos
Según el anterior razonamiento el espectro de IR de la molécula de agua debería de tener tres bandas de absorción. Tensión simétrica Tensión asimétrica Los espectros reales son más complejos: -sobretonos - bandas combinación. El agua en estado líquido da lugar a una gran cantidad de bandas combinación de pequeña intensidad Escala Logarítmica Espectro IR+microondas del vapor de agua

25
Espectro de agua líquida a T ambiente
698 nm

26
NH3. Moléculas no lineal. Modos de vibración = 3N-6 = 6
En realidad 4, ya que 2 dos degenerados Tensión simétrica Simetría a1 1 2 Aleteo Simetría a1 Flexión degenerada Simetría e Tensión asimétrica degenerada Simetría e 3 4

27
En disolución o en estado sólido desaparece la estructura rotacional de las bandas.

29
Espectroscopía electrónica.
En moléculas, las bandas que aparecen en espectroscopía electrónica son muy anchas, debido a que simultáneamente al salto electrónico tienen lugar saltos vibracionales y rotacionales. El principio de Franck-Condon nos dice que los saltos electrónicos tienen lugar a distancia interatómica constante. Supondremos una molécula diatómica, en su estado electrónico fundamental, S0, y en su estado vibracional fundamental, v=0, no considerándose, para simplificar, la estructura fina de rotación. La molécula absorbe radiación para pasar al estado electrónico excitado S1, con mínimo de energía a distancia interatómica mayor. Debido a que la distancia interatómica se mantiene constante durante el salto (Principio de Franck-Condon), la molécula se promociona hacia niveles excitados de vibración.
, la molécula se promociona hacia niveles excitados de vibración.")
30
Espectro de ClO gaseoso
Si el exceso de energía de vibración del estado electrónico excitado es suficiente, la molécula puede sufrir disociación (fotolisis o fotodisociación). Este fenómeno se observa nítidamente en el espectro gracias a la aparición de un continuo. Espectro de SO2 gaseoso Espectro de ClO gaseoso
. Este fenómeno se observa nítidamente en el espectro gracias a la aparición de un continuo. Espectro de SO2 gaseoso. Espectro de ClO gaseoso.")
32
Para moléculas poliatómicas
Para moléculas diatómicas, las reglas de selección son Para moléculas poliatómicas Hay que recordar que estas reglas no son inviolables, y solo nos indican si las transiciones son más o menos probables. Si se utiliza la aproximación de la valencia dirigida, los electrones de la molécula pueden clasificarse en , y n (no enlazantes) Transiciones permitidas (muy intensas) Transiciones parcialmente permitidas (débiles) Transiciones prohibidas
 Transiciones permitidas (muy intensas) Transiciones parcialmente permitidas (débiles) Transiciones prohibidas.")
33
Espectroscopía fotoelectrónica (UPS).
Se hacen incidir fotones monocromáticos de alta energía (h = constante) sobre la muestra, ionizandose el átomo o la molécula estudiada. Se analiza la energía cinética de los electrones desprendidos y se mide I = energía de ionización En función de la energía del fotón incidente, existen dos tipos de espectroscopias fotoelectrónicas. UPS, que utiliza radiación UV, e ioniza electrones de valencia (los más lábiles) XPS que utiliza rayos X y excita electrones internos (La espectroscopía Auger es una variante de la XPS). Como fuente de energía en UPS,se usa predominantemente la transición del He (58.4 nm)
 sobre la muestra, ionizandose el átomo o la molécula estudiada. Se analiza la energía cinética de los electrones desprendidos y se mide I = energía de ionización. En función de la energía del fotón incidente, existen dos tipos de espectroscopias fotoelectrónicas. UPS, que utiliza radiación UV, e ioniza electrones de valencia (los más lábiles) XPS que utiliza rayos X y excita electrones internos (La espectroscopía Auger es una variante de la XPS). Como fuente de energía en UPS,se usa predominantemente la transición del He (58.4 nm)")
34
Espectro UPS del Ar. 2P1/2 p5 2P3/2
La configuración p5 es equivalente a la p1. Para esta, L = 1 y S = 1/2, por lo que J = 3/2 y 1/2. Existen por lo tanto dos niveles de energía 2P3/2 y 2P1/2. El primer estado está 4 veces degenerado, 4 valores de MJ = 3/2, 1/2, -1/2 y -3/2. El segundo estado la degeneración es 2 ( MJ = 1/2 y -1/2). Por esta razón el área del primer pico es el doble que la del segundo.
. Por esta razón el área del primer pico es el doble que la del segundo.")
35
espectro UPS de la molécula de H2.
H2 + h →H2+ Las diferentes líneas del espectro corresponden a la estructura vibracional del estado electrónico fundamental del H2+.

36
el espectro UPS de del N2 N2+.
región A región B región C Se quita un e- de un OM antienlazante

37
Espectro UPS de del H2O H2O+.

38
Otros tipos de espectroscopía. Fluorescencia y Fosforescencia:
Supongamos que una molécula absorbe radiación y pasa del estado electrónico fundamental, S0, hasta el estado excitado, S1. Estos dos estados deben tener el mismo spin, ya que debe cumplirse que Supondremos además que S = 0 para los dos estados. Emisión espontánea S1 Tiempo del orden de 10-9 s. S0

39
Emisión espontánea S1 T1 Tiempo del orden de 10-3 -1 s. S0
Fluorescencia A* (S1)→ A + h’ Fosforescencia T1 A* (T1)→ A + h’’ Evolución del estado excitado Quenching (auto o medio) Tiempo del orden de s. A* → A A + h → A* Transferencia de enegía A* + B → A + B*→ A + B+h’ S0 Transferencia de carga A* + B → A+ + B Todos estos fenómenos compiten y su importancia relativa depende del medio y la temperatura
→ A + h’ Fosforescencia. T1. A* (T1)→ A + h’’ Evolución del estado excitado. Quenching (auto o medio) Tiempo del orden de s. A* → A. A + h → A* Transferencia de enegía. A* + B → A + B*→ A + B+h’ S0. Transferencia de carga. A* + B → A+ + B Todos estos fenómenos compiten y su importancia relativa depende del medio y la temperatura.")
40
Los espectros de emisión se pueden medir de tres formas diferentes
λ excitación = cte Espectros estacionarios de emisión Se excita a λ = cte y se representa el espectro en función de la λ de enisión λ de enisión Espectros estacionarios de excitación Se excita a λ = variable y se mide a λ = cte λ de enisión cte λ excitación Espectros de emisión de tiempo resuelto Se excita a λ = cte y se miden los fotones emitidos en función del tiempo sea cual sea λ de emisión

41
Transferencia de carga: Proceso fundamental para la construcción de células fotovoltáicas y dispositivos electroluminiscentes

42
Efecto Laser y Espectroscopia Laser:
Supongamos un sistema con un estado fundamental S0 y una pareja de estados excitados S1 y T1. Supongamos que la transferencia de S1 a T1 es muy alta y que las moléculas quedan bloqueadas durante algunos instantes antes de emitir la radiación de forma espontánea (fosforescencia). Se produce una inversión de población S1 T1 Transición permitida Transición prohibida S0 La emisión de radiación puede ser espontánea o inducida. Para que sea inducida, debemos comunicar al sistema radiación de energía idéntica a la de emisión. Cuando esto se hace adecuadamente se produce el efecto Laser (Light Amplification by Stimulated Emission of Radiation o aplificación de luz por emisión inducida de radiación). Cualquier sistema fosforescente puede ser usado para fabricar un laser.
. Se produce una inversión de población. S1. T1. Transición permitida. Transición prohibida. S0. La emisión de radiación puede ser espontánea o inducida. Para que sea inducida, debemos comunicar al sistema radiación de energía idéntica a la de emisión. Cuando esto se hace adecuadamente se produce el efecto Laser (Light Amplification by Stimulated Emission of Radiation o aplificación de luz por emisión inducida de radiación). Cualquier sistema fosforescente puede ser usado para fabricar un laser.")
43
Tipos de láseres: De estado sólido
El primer laser visible que se construyó fue el de cristales de rubí (1960) Cr3+ (4A2) es excitado con un flash de xenón hasta 4T2, desde el que se produce una transferencia de energía a un estado 2E1, El tiempo de vida media del estado 2E1, ~ s. Si se aplica antes de que transcurra este tiempo radiación apropiada, se produce la emisión inducida. Rubí: Al2O3 + Cr2O3( %). Se colocan unos espejos en los extremos del cristal, de forma que los primeros fotones emitidos por el rubí de forma espontánea, se reflejan en el espejo y pueden actuar induciendo la emisión de otros iones. Se deja un orificio en uno de los espejos por donde sale el rayo laser. Si es posible disipar el calor generado el laser trabaja en continuo, en caso contrario en pulsos. La radiación laser es muy intensa, monocromática y altamente direccionable. Tipos de láseres: De estado sólido
 Cr3+ (4A2) es excitado con un flash de xenón hasta 4T2, desde el que se produce una transferencia de energía a un estado 2E1, El tiempo de vida media del estado 2E1, ~ s. Si se aplica antes de que transcurra este tiempo radiación apropiada, se produce la emisión inducida. Rubí: Al2O3 + Cr2O3( %). Se colocan unos espejos en los extremos del cristal, de forma que los primeros fotones emitidos por el rubí de forma espontánea, se reflejan en el espejo y pueden actuar induciendo la emisión de otros iones. Se deja un orificio en uno de los espejos por donde sale el rayo laser. Si es posible disipar el calor generado el laser trabaja en continuo, en caso contrario en pulsos. La radiación laser es muy intensa, monocromática y altamente direccionable. Tipos de láseres: De estado sólido.")
44
Tipos de láseres: De excímeros
mezclas gaseosas de un gas noble (Xe, Ar o Kr) y un halógeno (F o Cl) y se hace pasar una descarga eléctrica, formándose moléculas del tipo XeCl* o XeF*, las cuales solo tienen existencia como estados excitados. El retorno al estado fundamental de estos sistemas es lento, lo que se utiliza para fabricar el laser.
 y un halógeno (F o Cl) y se hace pasar una descarga eléctrica, formándose moléculas del tipo XeCl* o XeF*, las cuales solo tienen existencia como estados excitados. El retorno al estado fundamental de estos sistemas es lento, lo que se utiliza para fabricar el laser.")
45
Tipos de láseres: De gases
Laser de Helio-Neon Laser de dióxido de carbono Laser de ión de Argón

46
Tipos de láseres: De colorantes (líquidos)
Se utilizan, en diferentes proporciones, una gran variedad de moléculas orgánicas que absorben en el visible. Con esto se pueden obtener lasers que emiten radiación a cualquier longitud de onda (laser continuo).
.")
47
Aplicaciones de los láseres en Química
Espectroscopía λ Muestra λ

48
Aplicaciones de los láseres en Química
Espectroscopía Espectro del benceno en etanol obtenidos mediante un espectrofotómetro convencional, en el que la fuente de radiación es una lámpara, y la radiación se hace pasar por un monocromador (un prisma por ejemplo). Los monocromadores no son perfectos y todos poseen una cierta anchura espectral. Espectro del benceno en etanol obtenidos mediante un espectrofotómetro en el que la fuente de radiación es un laser de colorantes. Espectro del benceno en estado gaseoso obtenidos mediante un espectrofotómetro en el que la fuente de radiación es un laser de colorantes.
. Los monocromadores no son perfectos y todos poseen una cierta anchura espectral. Espectro del benceno en etanol obtenidos mediante un espectrofotómetro en el que la fuente de radiación es un laser de colorantes. Espectro del benceno en estado gaseoso obtenidos mediante un espectrofotómetro en el que la fuente de radiación es un laser de colorantes.")
49
Otras aplicaciones de los láseres en Química
Separación de isótopos Fotodisociación selectiva de uno de los isótopos 1 A + h1+ h2 → A* Espectroscopía multifotónica Se excitan solo las moléculas del punto cofocal 2 Espectroscopía Raman Espectroscopía resuelta en el tiempo

Presentaciones similares




 ESTRUCTURA MOLECULAR>")















