Descargar la presentación
La descarga está en progreso. Por favor, espere

1
Diseño Molecular de Fármacos
Prof. Ramón Garduño Juárez Modelado Molecular Diseño de Fármacos

2
¿Qué es un modelo? Modelo Experimento Teoría
Un modelo sirve de puente entre la teoría y el experimento – en efecto un modelo llena las brechas en los datos experimentales.

3
¿Qué es un modelo molecular?
En sí, cualquier representación de una molécula es un modelo: Dibujos de líneas para estructuras orgánicas Modelos simples de “bolas y palitos” Mapas de densidad electrónica Representaciones Van der Waals o CPK Figuras de cilindros y listones para proteínas Estructuras cristalinas de rayos-X

4
Propiedades (importantes) observables
Moleculares Propiedades que están localizadas en una sola molécula Punto de fusión, logP, peso molecular, refractividad molar, HOMO, LUMO, etc. Algunas pueden ser ‘atomísticas’ Intermoleculares Propiedades que se derivan de la interacción de dos moléculas Constantes de unión, energía libre de unión, etc. No son muy ‘atomísticas’

5
Predicción de Propiedades
Las herramientas de cómputo pueden ser usadas para modelar propiedades. Más datos experimentales => mejores modelos y mejores predicciones. En general, las propiedades moleculares son más fáciles de predecir que las propiedades intermoleculares. Las herramientas de cómputo y los métodos para el descubrimiento de fármacos, están clasificados casi de igual manera.

6
Dos paradigmas en el descubrimiento de fármacos
Descubrimientos basados en el Ligando Usa las propiedades de las moléculas mismas para desarrollar modelos de actividad y asistir en el diseño de nuevos compuestos líderes activos y candidatos clínicos. Descubrimientos basados en la Estructura Usa las propiedades (y estructuras) de moléculas unidas a sus receptores para identificar las características moleculares ideales para unión y actividad. (LOCK and KEY).
 de moléculas unidas a sus receptores para identificar las características moleculares ideales para unión y actividad. (LOCK and KEY).")
7
Técnicas Clave del Modelado
Mecánica Cuántica Mecánica Molecular 3D QSAR Búsqueda en la Base de Datos de Moléculas Pequeñas Acoplamiento y tanteo Diseño de Fármacos De Novo

8
Mecánica Cuántica H Ψ = E Ψ
Estructura electrónica y propiedades, orbitales moleculares, energías de ionización, etc. Consideración es de tamaño y velocidad hacen a la MC casi imposible para proteínas, e impráctica para posibles moléculas de fármacos muy grandes.

9
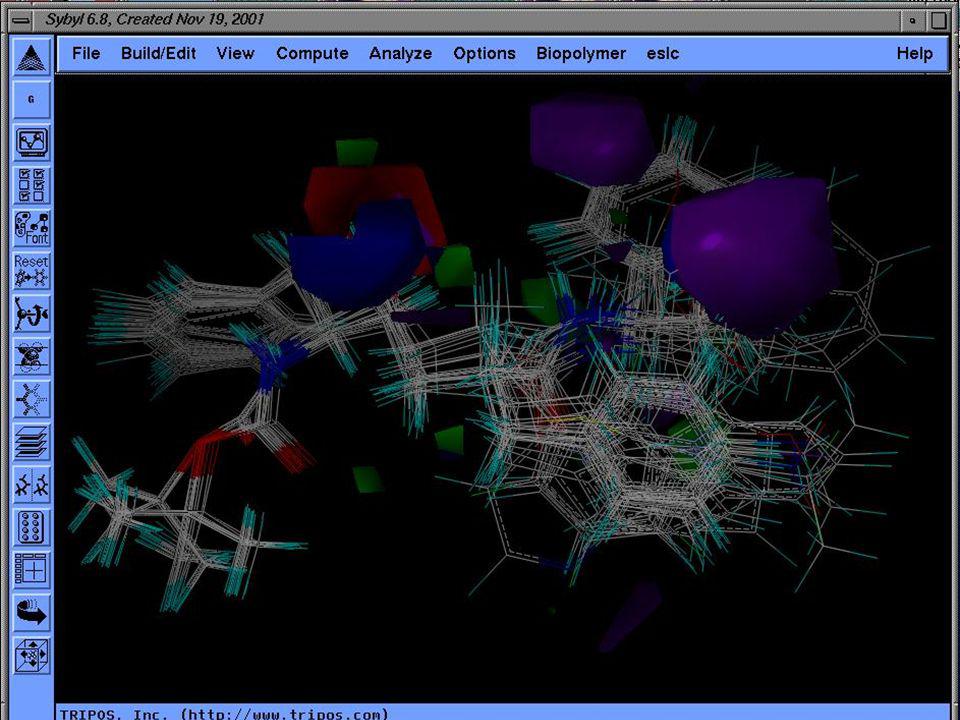
Mecánica Molecular

10
QSAR 3D (CoMFA)
")
11
Tabla QSAR

12
QSAR en 3-Dimensiones

13
Modelos QSAR 3D El uso de Mínimos Cuadrados Parciales (PLS) para derivar el modelo de actividad con una función de los valores puntuales de la malla.. La validación cruzada es crucial: indica la predictividad interna del modelo. Validar el modelo con un conjunto de prueba externa. Realizar los mapas de coeficientes en 3D => aprender la relación espacial entre estructura y actividad.
 para derivar el modelo de actividad con una función de los valores puntuales de la malla.. La validación cruzada es crucial: indica la predictividad interna del modelo. Validar el modelo con un conjunto de prueba externa. Realizar los mapas de coeficientes en 3D => aprender la relación espacial entre estructura y actividad.")
15
Búsquedas 3D en las Bases de Datos
La mayoría de las compañías farmacéuticas tienen bibliotecas privadas muy grandes de compuestos que han sido parte de proyectos anteriores. Muchos de éstos han sido aprobados contra tamizados particulares, pero no contra todos los tamizados El National Cancer Institute (NCI) tiene una biblioteca pública de > 300,000 compuestos. Algunas agencias comerciales venden otras bibliotecas con hasta 6,000,000 compuestos.
 tiene una biblioteca pública de > 300,000 compuestos. Algunas agencias comerciales venden otras bibliotecas con hasta 6,000,000 compuestos.")
16
Usando las Bases de Datos 3D
Un compuesto activo (competidor) se analiza para determinar su farmacóforo. Arreglos espaciales en 3D entre componentes del farmacóforo son usados para construir una pregunta. La pregunta se aplica a la base de datos 3D para derivar aciertos. Los aciertos se examinan por los químicos para determinar la posibilidad sintética, su novedad, etc. para generar las pistas.
 se analiza para determinar su farmacóforo. Arreglos espaciales en 3D entre componentes del farmacóforo son usados para construir una pregunta. La pregunta se aplica a la base de datos 3D para derivar aciertos. Los aciertos se examinan por los químicos para determinar la posibilidad sintética, su novedad, etc. para generar las pistas.")
17
Acoplamiento y Tanteo Colocar juntos los modelos moleculares del receptor y los ligandos putativos en 3-dimensiones “Tantear” las interacciones – i.e., predecir la unión y actividad Si se llevó a cabo sobre bases de datos muy grandes a esto algunas veces se le llama “Tamizado Virtual”

21
La Ecuación Maestra DG = DH - TDS

22
Hidrofobicidad Medida como el coeficiente de partición Agua / Octanol (P). log P > 0 : fase lípidos log P < 0 : fase agua

23
Método Leo (CLOG-P) i = número de ocurrencias del fragmento f de tipo n. j = número de ocurrencias del factor F de tipo m.

24
Hidropático Campo 1D LogP vs. 3D
El Log P de la base libre de cocaína es: 4.24 El Log P de la sal cuaternaria de cocaína es: 1.10 Sin embargo, si examinamos los mapas hidropáticos en 3D para estas dos moléculas, podemos ver perfiles muy diferentes de cómo la cocaína se puede presentar a sí misma como un ligando

25
Mapa Hidropático 3D para la Sal cuaternaria de Cocaína

26
Mapa Hidropático 3D para la Base Libre de Cocaína

27
B = SS bij = SS aiSi ajSj Rij Tij + rij
La Ecuación “HINT B = SS bij = SS aiSi ajSj Rij Tij + rij a = constante atómica hidrofóbica S = área de superficie accesible al disolvente Rij = exponencial (e-r) Tij = función de discriminante para interacciones polar-polar rij = término de van der Waals
 Tij = función de discriminante para interacciones polar-polar. rij = término de van der Waals.")
28
Clases de interacciones No Covalentes
Puentes de hidrógeno - A-H…B- 1 – 10 kcal/mol Coulombicas -Ad+ Bd-- 0.5 – 5 kcal/mol Hidrofóbicas -CHn CHm- 0.5 – 2 kcal/mol Van der Waals (London) -X Y- < 1 kcal/mol Nota: cualquier método comprensivo que intente el modelar la unión del ligando también debe considerar la energía de solvatación y contribuciones entropicas al proceso de unión.
 -X Y- < 1 kcal/mol. Nota: cualquier método comprensivo que intente el modelar la. unión del ligando también debe considerar la energía de. solvatación y contribuciones entropicas al proceso de unión.")
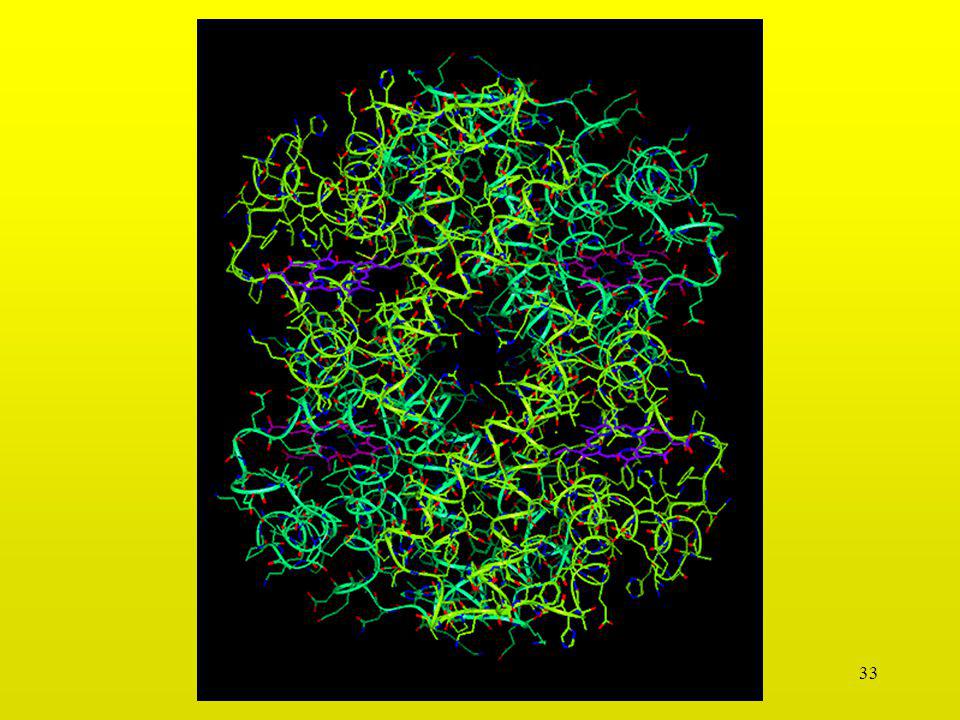
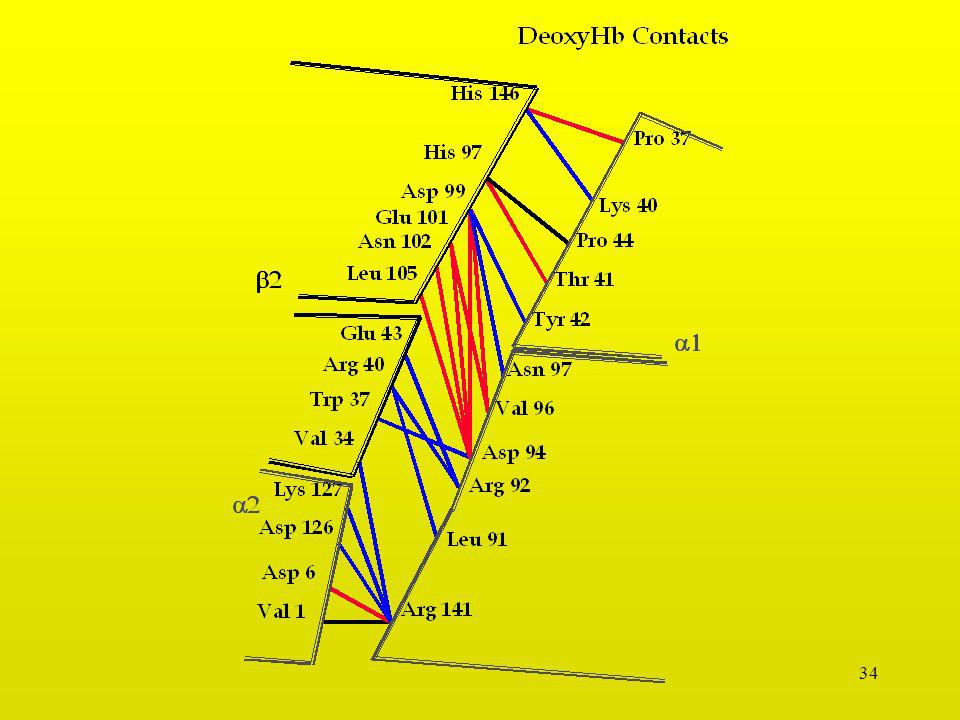
29
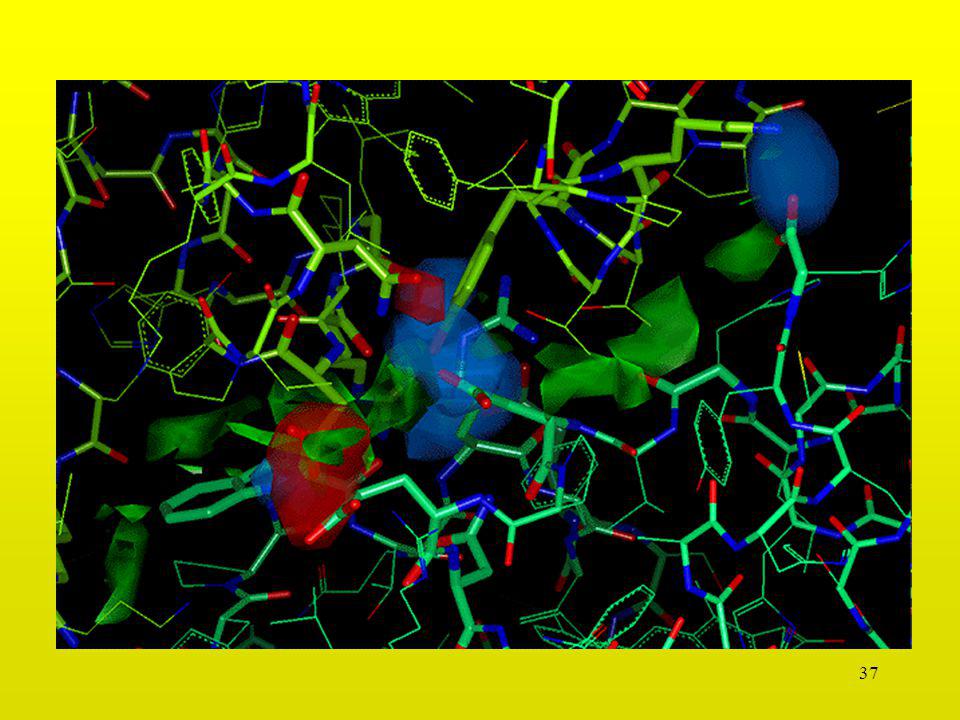
Interacción Serina-Glicina

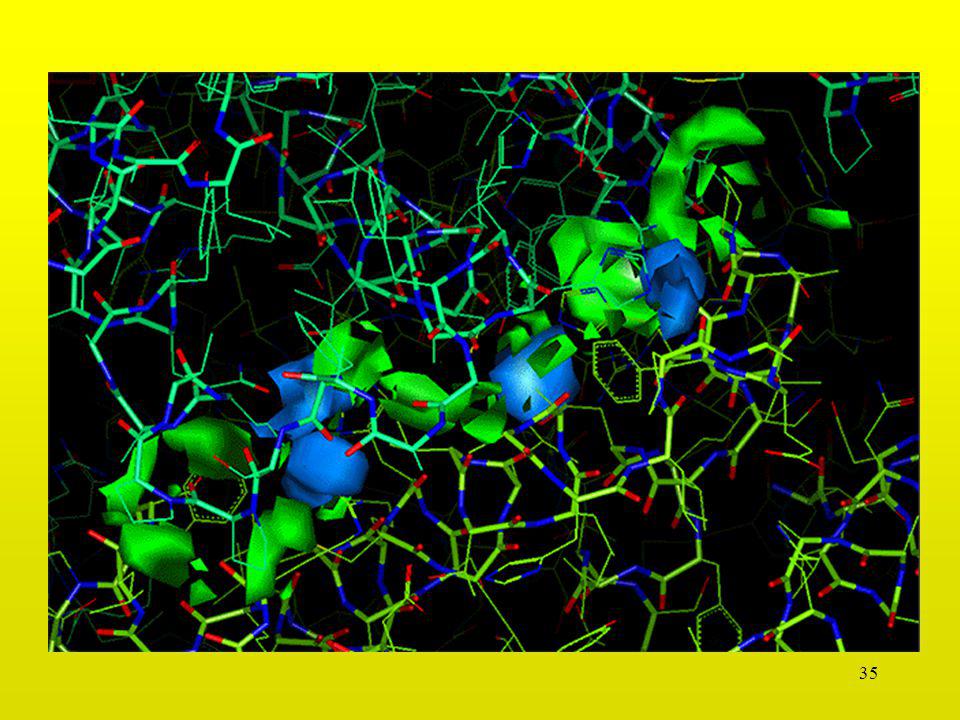
30
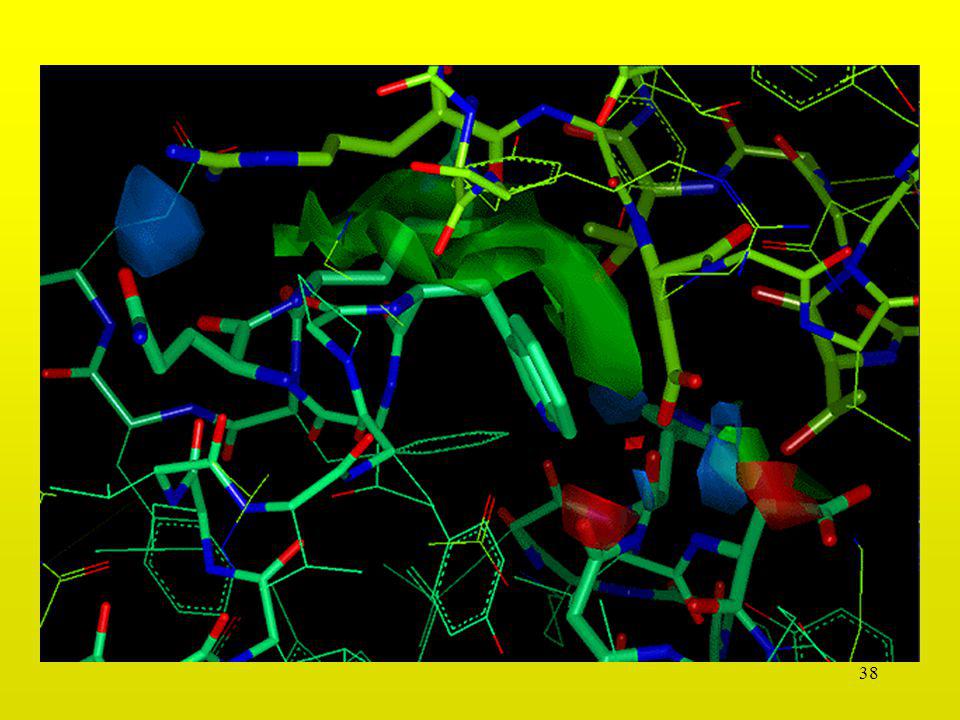
Interacción Asp-Asp

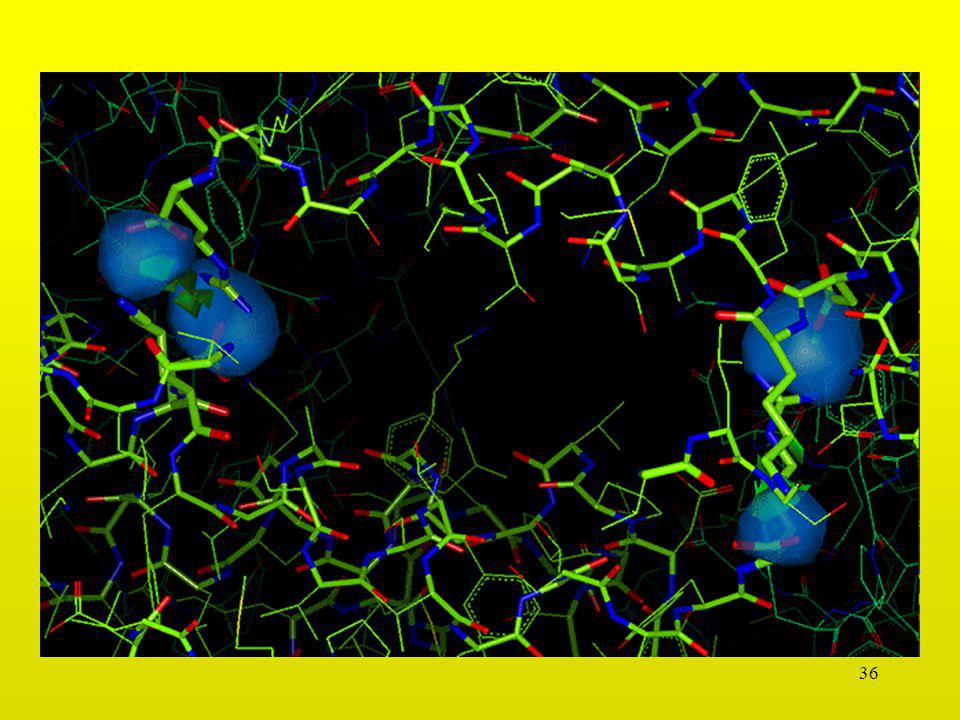
31
Interacción Leu-Leu

39
Energía libre de unión del ligando

40
Ecuaciones HINT “QSAR” para DG:
DG = HTOTAL – kcal mol-1 (error estándar = ±2.6 kcal mol-1, r = 0.54) i.e., – 513 HINT unidades / kcal mol-1 or DG = – (HHB + HAB) – HHH – HAA – HBB – HHP – kcal mol-1 (error estándar = ±1.8 kcal mol-1, r = 0.82)
 i.e., – 513 HINT unidades / kcal mol-1. or. DG = – (HHB + HAB) – HHH. – HAA – HBB. – HHP – kcal mol-1. (error estándar = ±1.8 kcal mol-1, r = 0.82)")
42
¿Que hay del agua & solvatación?
El agua no es un simple “espectador” a la unión del ligando o a las asociaciones proteina-proteina! En general el agua puede ser el componente más importante del sistema: Cambios entropía surgen principalmente de los cambios en la estructura del agua Energías de solvatacion/desolvatación son causadas por cambios en la estructura de agua/biomolecula y agua/ligando La estructura del agua es difícil de observar experimentalmente o de predecir. Las estructuras de rayos-X no siempre pueden resolverse para el agua u otros iones.

43
L R Total = R-L + L-H2O [+ R-H2O]
![L R Total = R-L + L-H2O [+ R-H2O]](http://slideplayer.es/slide/1721609/7/images/43/L+R+Total+%3D+R-L+%2B+L-H2O+%5B%2B+R-H2O%5D.jpg "L R Total = R-L + L-H2O [+ R-H2O]")
44
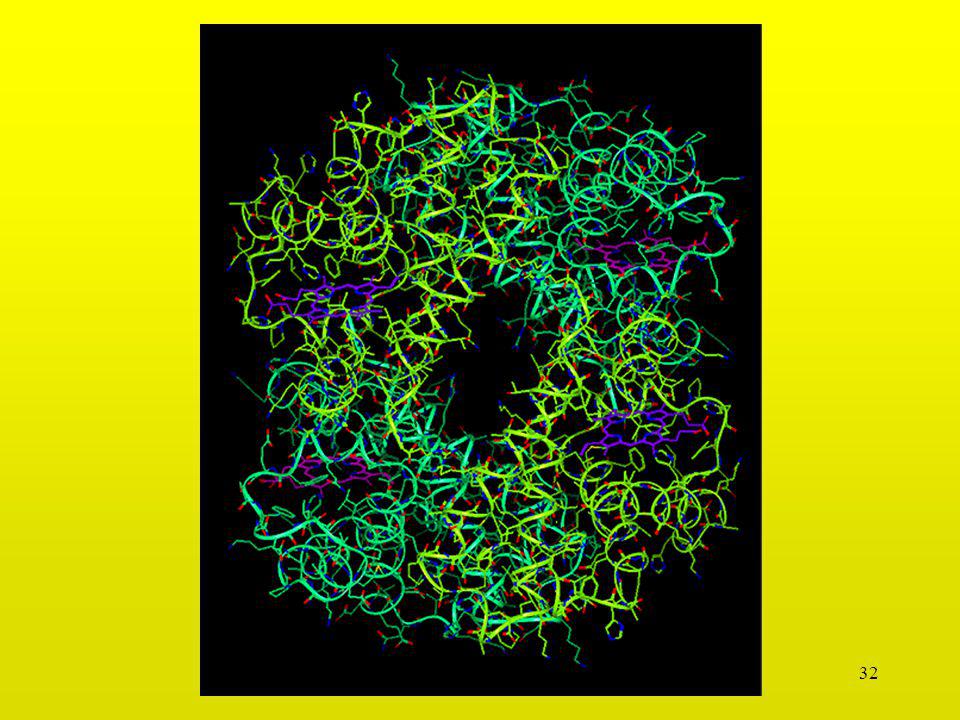
Proteasa HIV-1 (sin complejar)
D29 R108 wat300 wat301 D25 D125 I150 I50 R8 R187 D129

45
Proteasa HIV-1(complejada)
wat301 wat313bis’ wat313’ wat313bis wat313 D25 D125 I150 I50 R108 R8 D29 R187 R87 D129

46
Correlación de Tanteo HINT(sin agua)
n = 23 complejos HIV-1 proteasa-ligando ∆G = HTOTAL – 7.903 r = 0.55, Std. Error = ± 1.30 ∆G (kcal mol-1) HINT score
 HINT score.")
47
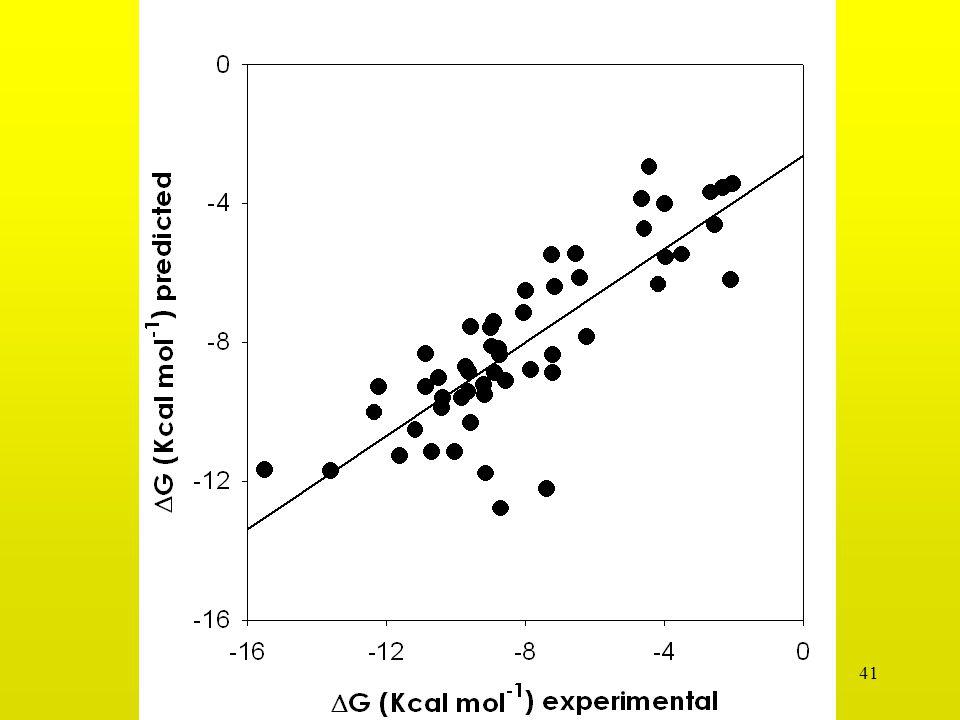
Correlación de Tanteo HINT (incl. 301 aguas)
n = 23 complejos HIV-1 proteasa-ligando ∆G = HTOTAL – 4.789 r = 0.79, Std. Error = ± 0.95 ∆G (kcal mol-1) HINT score
 HINT score.")
48
Por último El Diseño de Fármacos De Novo
Quizá el enfoque más especulativo Básicamente permite a las computadoras diseñar moléculas basadas en conjuntos de reglas químicas, diccionarios y patrones. Puede involucrar algoritmos genéticos u otras matemáticas sofisticadas. Las moléculas son “clasificadas” conforme se construyen y aquellas con la mejor clasificación son reportadas.

Presentaciones similares













 Noviembre de 2004.>")
 Noviembre de 2004.>")
 29 de julio de 2004.>")




