Descargar la presentación
La descarga está en progreso. Por favor, espere

1
Similaridad de cadenas genéticas Bienvenido Martínez Redondo Sergio García Esteban 2008/2009

2
Filogenia y similaridad de cadenas Estructura del ADN –Una molécula de ADN esta formada por dos cadenas, llamadas nucleótidos. –Cada nucleótido esta formado por una molécula de azúcar, un grupo fosfato y un compuesto nitrogenado llamado base. –Esta base puede ser adenina(A), citosina(C), guanina(G) o timina(T) –Las cadenas de genes vendrán representadas por una serie de letras A-C-G-T.
, citosina(C), guanina(G) o timina(T) –Las cadenas de genes vendrán representadas por una serie de letras A-C-G-T..")
3
Filogenia y similaridad de cadenas Mutaciones –Un cambio en alguno de los nucleótidos de una molécula de ADN provoca una mutación. –Las mutaciones son la única fuerza evolutiva capaz de crear variabilidad. –Existen dos tipos de mutaciones: Las producidas por la inserción o deleción de un nucleótido. Las producidas por la sustitución de un nucleótido por otro.

4
Filogenia y similaridad de cadenas Filogenia –El objetivo de la filogenia es convertir la información de las secuencias de nucleótidos en un árbol evolutivo de las mismas. –El proceso consta de dos pasos: Primero, alineamiento de secuencias. Segundo, construcción del árbol.

5
Filogenia y similaridad de cadenas Alineamiento de secuencias –El alineamiento de secuencias consiste en comparar dos secuencias homólogas para determinar la divergencia desde un ancestro común. –Cuando dos secuencias queden alineadas, se producirán tres tipos de emparejamientos: Matches: coincidencias en el mismo lugar del mismo nucleótido. Mismatches: no coincidencias en el mismo lugar del mismo nucleótido. Gaps: son emparejados entre “nada” y el nucleótido. Se producen por el proceso de alineamiento.

6
Filogenia y similaridad de cadenas Alineamiento de secuencias –Existen dos tipos de alineamiento: Local: Buscan regiones entre dos proteínas que son parecidas, aunque se hallen rodeadas de zonas completamente diferentes. Útiles para proteínas modulares. Global: Se extiende a lo largo de toda la longitud de las secuencias. Útiles cuando las secuencias son parecidas a lo largo de toda su longitud.

7
Algoritmos para el alineamiento global Matriz de Puntos (Gibss & McIntyre) –Consiste en colocar una de las cadenas en la primera fila y la otra en la primera columna, marcando los puntos en los que coincidan. –Después se trata de trazar rectas que unan de la mejor forma posible, la esquina superior izquierda con la esquina inferior derecha, de forma que pasemos por el mayor número de puntos.

8
Filogenia y similaridad de cadenas Algoritmo de Needleman-Wunsch –Fue desarrollado en 1970 por Saul Needleman y Chistian Wunsch. –Esta basado en programación dinámica y siempre encuentra la solución óptima. –El proceso que sigue es el siguiente: Cadenas a alinear son A y B, con |A|=m,|B|=n. Definimos la matriz S, donde cada elemento S(i,j) indica la similitud entre los simbolos el alfabeto. Definimos también una variable d que indica la penalización por hueco.
 indica la similitud entre los simbolos el alfabeto. Definimos también una variable d que indica la penalización por hueco..")
9
Filogenia y similaridad de cadenas Algoritmo de Needleman-Wunsch Creamos una matriz interna llamada F, que almacenara los resultados parciales de cada alineamiento. En cada iteración, el valor (i,j) de F, indica el alineamiento óptimo entre A[0,i] y B[0,i]. Por tanto el elemento (n,m) de F contendrá el alineamiento óptimo. Una vez calculada F, solo queda obtener la secuencia de alineamiento consistente en llegar desde la posición (n,m) hasta la (1,1)
 de F, indica el alineamiento óptimo entre A[0,i] y B[0,i]. Por tanto el elemento (n,m) de F contendrá el alineamiento óptimo. Una vez calculada F, solo queda obtener la secuencia de alineamiento consistente en llegar desde la posición (n,m) hasta la (1,1).")
10
Filogenia y similaridad de cadenas Algoritmo de Needleman-Wunsch –Explicación de la matriz S: Para estas secuencias: AGCCTATC ACC_T_TC ACC_T_TC Con d= -5 Con S= Tendríamos: Score = S(A,A) + S(G,C) + S(C,C) + d + S(T,T) + d + S(T,T) + S(C,C) = 29 AGCT A10-3-4 G7-5-3 C-3-590 T-4-308
 + S(G,C) + S(C,C) + d + S(T,T) + d + S(T,T) + S(C,C) = 29 AGCT A G7-5-3 C T-4-308")
11
Filogenia y similaridad de cadenas Algoritmo de Needleman-Wunsch Inicio del algoritmo: F0j = d * j Fi0 = d * i Recursión para obtener el siguiente elemento de forma óptima: Fij = max(Fi − 1,j − 1 + S(Ai,Bj),Fi,j − 1 + d,Fi − 1,j + d) Calculamos la matriz F: for i=0 to length(A)-1 F(i,0) <- d*i for j=0 to length(B)-1 F(0,j) <- d*j for i=1 to length(A) for j = 1 to length(B) { Choice1 <- F(i-1,j-1) + S(A(i), B(j)) Choice1 <- F(i-1,j-1) + S(A(i), B(j)) Choice2 <- F(i-1, j) + d Choice2 <- F(i-1, j) + d Choice3 <- F(i, j-1) + d Choice3 <- F(i, j-1) + d F(i,j) <- max(Choice1, Choice2, Choice3) } F(i,j) <- max(Choice1, Choice2, Choice3) }
,Fi,j − 1 + d,Fi − 1,j + d) Calculamos la matriz F: for i=0 to length(A)-1 F(i,0) <- d*i for j=0 to length(B)-1 F(0,j) <- d*j for i=1 to length(A) for j = 1 to length(B) { Choice1 <- F(i-1,j-1) + S(A(i), B(j)) Choice1 <- F(i-1,j-1) + S(A(i), B(j)) Choice2 <- F(i-1, j) + d Choice2 <- F(i-1, j) + d Choice3 <- F(i, j-1) + d Choice3 <- F(i, j-1) + d F(i,j) <- max(Choice1, Choice2, Choice3) } F(i,j) <- max(Choice1, Choice2, Choice3) }")
12
Filogenia y similaridad de cadenas Algoritmo de Needleman-Wunsch AlignmentA <- "" AlignmentB <- "" AlignmentA <- "" AlignmentB <- "" i <- length(A) - 1 j <- length(B) – 1 while (i > 0 AND j > 0) { while (i > 0 AND j > 0) { Score <- F(i,j) ScoreDiag <- F(i - 1, j - 1) ScoreUp <- F(i, j - 1) ScoreLeft <- F(i - 1, j) if (Score == ScoreDiag + S(A(i), B(j))) { AlignmentA <- A(i-1) + AlignmentA AlignmentB <- B(j-1) + AlignmentB i <- i - 1 j <- j - 1 } else if (Score == ScoreLeft + d) { AlignmentA <- A(i-1) + AlignmentA AlignmentB <- "-" + AlignmentB i <- i - 1 } otherwise (Score == ScoreUp + d) { AlignmentA <- "-" + AlignmentA AlignmentB <- B(j-1) + AlignmentB j <- j - 1 } } while (i > 0) { while (i > 0) { AlignmentA <- A(i-1) + AlignmentA AlignmentB <- "-" + AlignmentB i <- i - 1 } while (j > 0) { AlignmentA 0) { AlignmentA <- "-" + AlignmentA AlignmentB <- B(j-1) + AlignmentB j <- j - 1 }
 - 1 j <- length(B) – 1 while (i > 0 AND j > 0) { while (i > 0 AND j > 0) { Score <- F(i,j) ScoreDiag <- F(i - 1, j - 1) ScoreUp <- F(i, j - 1) ScoreLeft <- F(i - 1, j) if (Score == ScoreDiag + S(A(i), B(j))) { AlignmentA <- A(i-1) + AlignmentA AlignmentB <- B(j-1) + AlignmentB i <- i - 1 j <- j - 1 } else if (Score == ScoreLeft + d) { AlignmentA <- A(i-1) + AlignmentA AlignmentB <- - + AlignmentB i <- i - 1 } otherwise (Score == ScoreUp + d) { AlignmentA <- - + AlignmentA AlignmentB <- B(j-1) + AlignmentB j <- j - 1 } } while (i > 0) { while (i > 0) { AlignmentA <- A(i-1) + AlignmentA AlignmentB <- - + AlignmentB i <- i - 1 } while (j > 0) { AlignmentA 0) { AlignmentA <- - + AlignmentA AlignmentB <- B(j-1) + AlignmentB j <- j - 1 }")
13
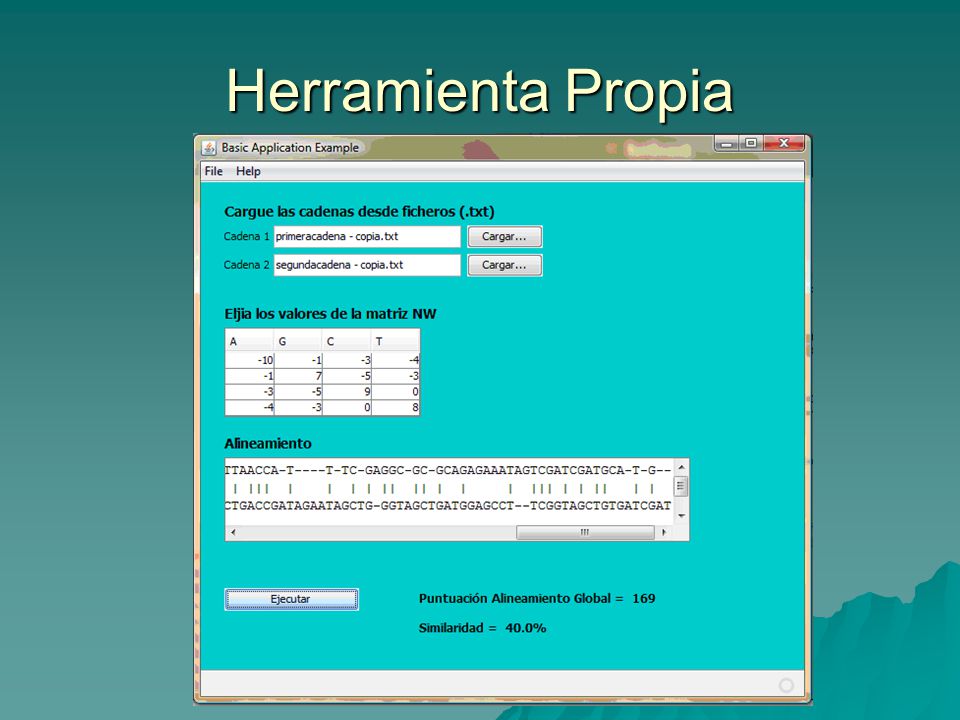
Herramienta Propia –Hemos desarrollado una herramienta, que implementa el algoritmo de Needleman- Wunsch. –La hemos implementado en java y desarrollado con el NetBeans. –Esta herramienta nos ofrece la posibilidad de cargar las cadenas mediante ficheros, modificar la matriz S y nos muestra tanto el alineamiento de las cadenas como las puntuaciones de similaridad. –En la dirección http://xylian.igh.cnrs.fr/ se puede encontrar una herramienta llamada ALIGN muy parecida. http://xylian.igh.cnrs.fr/

14
Herramienta Propia

16
Conclusiones -Amplitud del campo de la BioInformática -Conseguir abstraer los problemas puramente técnicos -Nuestro tema se planteaba muy bien desde un punto de vista informático -Necesidad de comunicación entre ambos sectores

Presentaciones similares










>")




.>")



