Descargar la presentación
La descarga está en progreso. Por favor, espere

1
SÍNDROMES MIASTENIFORMES
JEFE CURSO: DR. ENRIQUE JUAN DÍAZ GREENE PROFESOR ADJUNTO: DR. FEDERICO LEOPOLDO RODRÍGUEZ WEBER SUPERVISÓ: DR. ARTURO VIOLANTE PRESENTA: DRA. GRETA REYES R2MI

2
RESEÑA HISTÓRICA Thomas Willis (1621-1675) Médico Inglés, Oxford
Primero en describir lo hoy conocido como Miastenia gravis, 1672. Además describió el Polígono de Willis Rev Neurocir 2001; IV(4) :
 :")
3
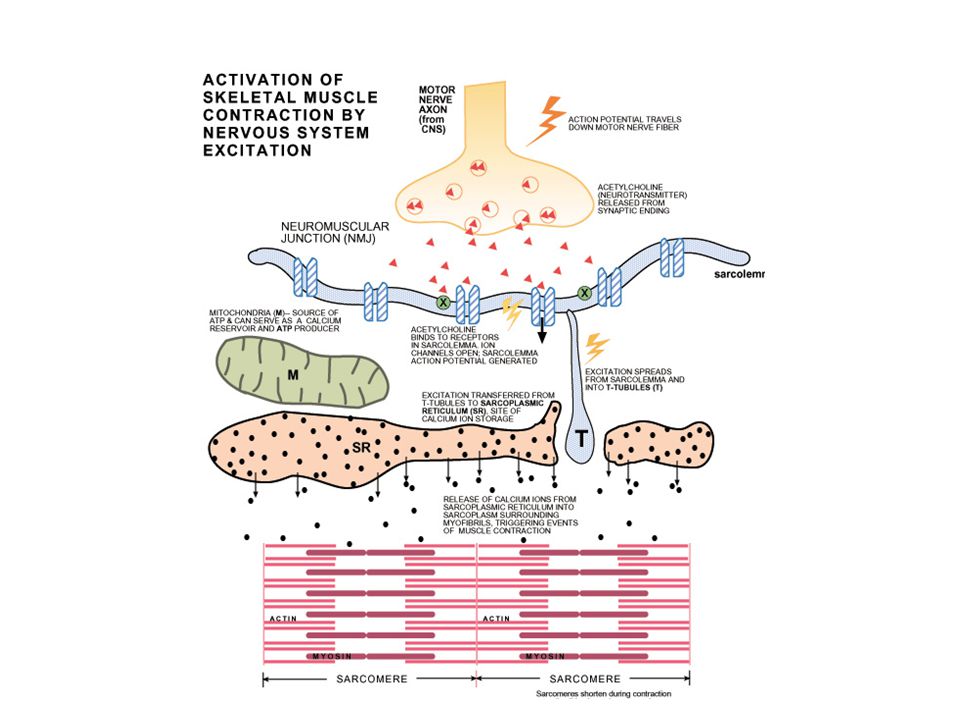
DEFINICIÓN Aquellos desordenes que afectan la normal función de la placa neuromuscular. Lancet 2001; 357: 2122–28

4
DESORDENES DE LA UNIÓN NEUROMUSCULAR
Congénitos y adquiridos. Adquiridos: Botulismo Miastenia gravis: Autoinmune*** Inducida por medicamentos *** Neonatal: transitoria Ocular Timoma Síndrome miasténico “Eaton Lambert” Toxinas por veneno de serpientes

6
EATON LAMBERT BOTULISMO MIASTENIA TOXINAS VENENO

7
EPIDEMIOLOGÍA Miastenia gravis Incidencia: 10-20 casos/ millón
Prevalencia: 1/20,000 Mujeres 3x, años Hombres: 60 años 600 pacientes en H. Siglo XXI Semin Neurol. 2004;24(1):17. J Am Geriatr Soc 2000 Nov;48(11):1442
:17. J Am Geriatr Soc 2000 Nov;48(11):1442.")
8
ETIOLOGÍA Enfermedad autoinmune que afecta receptores nicotínicos de acetilcolina en la fibra postsináptica. Desorden heterogéneo con alta predisposición genética. Lancet 2001; 357: 2122–28

9
ETIOLOGÍA La causa conocida más importante es la presencia de un timoma. El % persona con un timoma desarrolla Miastenia gravis. 10% personas con Miastenia gravis, tiene un timoma. 1-2% desarrolla Miastenia gravis bajo tratamiento con Penicilamina. Grob D. Natural history of myasthenia gravis. In: Engel AG, ed. Myasthenia gravis and myasthenic disorders. Oxford: Oxford University Press. Contemporary Neurology Series, 1999: 131–45.

10
FISIOPATOLOGÍA Tres mecanismos de daño en placa neuromuscular en fibra postsináptica: 1.- Daño por lisis mediante reacción por complemento. ** 2.- Anticuerpos propiamente: unión con receptores bloqueando y destruyendo el receptor. 3.- En algunos pacientes solo como bloqueador del receptor. **Mecanismo de daño más importante para determinar el grado de daño. Drachman DB, Adams RN, Josifek LF, Self SG. Functional activities of autoantibodies to acetylcholine receptors and the clinical severity of myasthenia gravis. N Engl J Med 1982; 307: 769–75.

11
Respecto al Timoma ( epitelial): Existen células T y B en centros germinales. Las células T estimulan a las células B las cuales producen anticuerpos contra ACTr. No se ha encontrado la relación, se cree que las células epiteliales tiene epítopes que comparte el receptor de acetilcolina. Las células epiteliales sensibilizan a las células T respecto al epítope y así se desencadene la producción de anticuerpos. Marx A, Schultz A, Wilisch A, Helmreich M, Nenninger R, Muller- Hermelink HK. Paraneoplastic autoimmunity in thymus tumors. Dev Immunol 1998; 6: 129–40.

12
CUADRO CLÍNICO Inicio insidioso o subagudo
Frecuentemente precedido por un factor desencadenante: fiebre, infección, cirugía, embarazo, parto, situaciones de agotamiento físico o emocional, ejercicio intenso o el uso de fármacos como aminoglucósidos o β-bloqueadores Puede ocurrir, aunque es infrecuente, el debut en forma de crisis miasténica.

13
CUADRO CLÍNICO No hay dolor Fatiga y debilidad (fluctuante)
Debilidad marcada ante actividad física Topografía: variada Ptosis y diplopia ** Alteraciones en la fonación, deglución, masticación. Menor expresión facial Disminución fuerza cervical, caía de cabeza o difícil sostén. Disminución en extremidades, músculos proximales (superiores++, inferiores+) Músculos respiratorios: monitorización mediante CVF No repercusión en reflejos osteotendinosos ni sensitivos Muscle Nerve. 2008;37(2):141.
 Músculos respiratorios: monitorización mediante CVF. No repercusión en reflejos osteotendinosos ni sensitivos. Muscle Nerve. 2008;37(2):141.")
14
CUADRO CLÍNICO Debilidad fluctuante. 80% tienen involucro ocular.
25% con diplopia, 25% con ptosis. 15% bulbar 5% extremidades ( solamente) 1 % falla respiratoria. El 50& de los qu einican con manifestaciones oculares, en 2 años desarrollarán miastenia generalizada Acta Neurol Scand. 2003;108(3):209.
 1 % falla respiratoria. El 50& de los qu einican con manifestaciones oculares, en 2 años desarrollarán miastenia generalizada. Acta Neurol Scand. 2003;108(3):209.")
15
EVOLUCIÓN El 85% de las MG ocular desarrollan debilidad generalizada en 1-2 años, progresa La MG se mantiene ocular en 15-20%. La remisión espontánea ocurre en 10 a 15% a los 2 años. A los 4 años se estabiliza su curso clínico.

16
Clasificación de Osserman modificada
Estadio I Miastenia ocular 20% Estadio IIa Miastenia generalizada leve; lenta progresión, sin crisis, con buena respuesta farmacológica 30% Estadio IIb Miastenia generalizada moderada; afectación muscular periférica y bulbar sin crisis. Poca respuesta farmacológica 20% Estadio III Crisis miasténica fulminante, progresión rápida, respuesta pobre a fármacos, insuficiencia respiratoria, alta incidencia de timoma mortalidad elevada 11% Estadio IV Miastenia de aparición tardía, transcurren 2 años en progresar desde I ó II a III, crisis respiratorias y mala respuesta a fármacos. Mortalidad elevada 9% Chinese Medical Journal 2010;123(18):
:")
17
DIAGNÓSTICO Clínica, se confirma. Dos pruebas
Prueba de hielo: consiste en el enfriamiento de la musculatura orbitaria con una bolsa de hielo sobre el ojo cerrado durante 2 min. Se considera positiva cuando la hendidura palpebral aumenta 2 mm o más respecto a la situación basal. La primera descripción que se conoce de la Miastenia Gravis fue realizada en 1672 por Thomas Willis, un médico y profesor de historia natural de Oxford, que todos conocemos más por ser el primer poligonero de la anatomía humana (en otras palabras, por ser el descubridor del Polígono de Willis). Tuvieron que pasar más de 200 años desde aquella primera descripción para que el alemán W.Heinrich Erb dijera que los casos documentados hasta el momento de extrañas parálisis bulbares similares al descrito por Willis, tal vez, fueran otra enfermedad. En 1878, presentó un informe en el que diferenciaba, en esta nueva forma de parálisis bulbar, tres características típicas: la caída bilateral de los párpados, la debilidad grave del cuello y los problemas masticatorios. La enfermedad toma el nombre de Síndrome de Erb-Goldflam, después de que Samule Goldfalm, un médico polaco, fundador de una policlínica para pacientes pobres con enfermedades neurológicas, describiera en 1893 la mejoría parcial de varios casos de la nueva parálisis bulbar. La primera vez que la enfermedad fue llamada Miastenia Gravis fue en Friedrich Jolly describe, en una joven paciente de 14 años, una rápida pero reversible fatiga de las extremidades,así como una fatigabilidad de la musculatura de las piernas. Jolly pensó que la causa podría estar en el músculo y no en el bulbo. De ahí, el nombre: mio = músculo; astenia = debilidad; gravis = intensa. Hace, aproximadamente, unos veinte años, el descubrimiento del déficit de los receptores de acetilcolina en la unión neuromuscular dio esperanza a los enfermos de Miastenia Gravis. Esta alteración de la placa neuromuscular, adquirida y autoinmune, era producida por anticuerpos dirigidos contra los receptores nicotínicos de acetilcolina situados en la membrana postsináptica de la placa muscular. Las formas fatales de otros tiempos pueden ser tratadas de forma efectiva y casi todos los pacientes pueden desarrollar una vida normal y activa. Eso quiere decir que los pacientes afectos de Miastenia Gravis llegan al quirófano programado en mayor cantidad que en el pasado. Y debemos estar preparados para anestesiarlos. He aquí mi revisión sobre el tema, que os podéis descargar. Kubis KC, Danesh-Meyer HV, Savino PJ, Sergott RC. The ice test versus the rest in myasthenia gravis. Ophthalmology 2000; 107:
. Tuvieron que pasar más de 200 años desde aquella primera descripción para que el alemán W.Heinrich Erb dijera que los casos documentados hasta el momento de extrañas parálisis bulbares similares al descrito por Willis, tal vez, fueran otra enfermedad. En 1878, presentó un informe en el que diferenciaba, en esta nueva forma de parálisis bulbar, tres características típicas: la caída bilateral de los párpados, la debilidad grave del cuello y los problemas masticatorios. La enfermedad toma el nombre de Síndrome de Erb-Goldflam, después de que Samule Goldfalm, un médico polaco, fundador de una policlínica para pacientes pobres con enfermedades neurológicas, describiera en 1893 la mejoría parcial de varios casos de la nueva parálisis bulbar. La primera vez que la enfermedad fue llamada Miastenia Gravis fue en Friedrich Jolly describe, en una joven paciente de 14 años, una rápida pero reversible fatiga de las extremidades,así como una fatigabilidad de la musculatura de las piernas. Jolly pensó que la causa podría estar en el músculo y no en el bulbo. De ahí, el nombre: mio = músculo; astenia = debilidad; gravis = intensa. Hace, aproximadamente, unos veinte años, el descubrimiento del déficit de los receptores de acetilcolina en la unión neuromuscular dio esperanza a los enfermos de Miastenia Gravis. Esta alteración de la placa neuromuscular, adquirida y autoinmune, era producida por anticuerpos dirigidos contra los receptores nicotínicos de acetilcolina situados en la membrana postsináptica de la placa muscular. Las formas fatales de otros tiempos pueden ser tratadas de forma efectiva y casi todos los pacientes pueden desarrollar una vida normal y activa. Eso quiere decir que los pacientes afectos de Miastenia Gravis llegan al quirófano programado en mayor cantidad que en el pasado. Y debemos estar preparados para anestesiarlos. He aquí mi revisión sobre el tema, que os podéis descargar. Kubis KC, Danesh-Meyer HV, Savino PJ, Sergott RC. The ice test versus. the rest in myasthenia gravis. Ophthalmology 2000; 107:")
18
Tensilon ( edrofonio): 2mg, 60s-2mg Iv, max 10 mg.
Inhibidor de la acetilcolinesterasa, aumenta los niveles de ACT y permite mayor duración del estímulo. Tras movilización repetitiva de una extremidad y ante fatiga se administra, con mejoría en la fuerza muscular. Duración 5 minutos Preparado siempre: Atropina para revertir posibles efectos adversos ( nauseas, vómito, sudoración, salivación) Precaución de hipotensión, falla respiratoria. (S:86-95%/E: 80-95%) Semin Neurol. 2004;24(1):31.
 Precaución de hipotensión, falla respiratoria. (S:86-95%/E: 80-95%) Semin Neurol. 2004;24(1):31.")
19
DIAGNÓSTICO Anticuerpos anti-receptor de acetilcolina.
S: 90% MG general/65% MG Ocular. Especificidad: 99% 60% ocular, 80% generalizada leve, 90% severa y 70% en remisión clínica. Anticuerpos MuSK 38-50% 6-12 % son seronegativos Muscle Nerve. 1992;15(6):720.
:720.")
20
DIAGNÓSTICO Electrofisiología : estimulación repetitiva ( Jolly) Se estimula eléctricamente un nervio de un musculo de 6-10 veces ( 2-3 Hertz). En el potencial de acción normal no hay cambio en la amplitud. En MG descenso progresivo en los primeros 4-5 estímulos. Estudio con sensibilidad de 75% Positivo: decremento en 10% Semin Neurol. 2004;24(1):31.
 Se estimula eléctricamente un nervio de un musculo de 6-10 veces ( 2-3 Hertz). En el potencial de acción normal no hay cambio en la amplitud. En MG descenso progresivo en los primeros 4-5 estímulos. Estudio con sensibilidad de 75% Positivo: decremento en 10% Semin Neurol. 2004;24(1):31.")
21
DIAGNÓSTICO Electromiografía:
Colocación de dos electrodos en dos músculos inervados por una misma neurona motora. Se estimula la neurona y se mide el tiempo del potencial de acción. Se denomina Jitter el parámetro valorado, el cual consiste en el cálculo las variaciones de los tiempos de transmisión neuromuscular en las descargas sucesivas Velocidad normal: 55 milisegundos para musculo extensor común de los dedos, 45 milisegundos para musculo frontal. Sensibilidad de estudio 95% J Neurol Neurosurg Psychiatry 1986;46: 677‑85

22
DIAGNÓSTICO TAC o RMN Torácica, en búsqueda de timoma.
75% pacientes con MG tiene alguna anormalidad tímica. 85% pacientes tienen hiperplasia tímica 15% timoma N Engl J Med. 1994;330(25):1797.
:1797.")
23
COMORBILIDADES 3-8 % personas con alguna alteración autoinmune tiroidea presente MG Colagenopatía (LEG, AR), anemia perniciosa 5% de los casos de MG N Engl J Med. 1994;330(25):1797.
, anemia perniciosa 5% de los casos de MG. N Engl J Med. 1994;330(25):1797.")
24
DIAGNÓSTICO DIFERENCIAL
Oftalmopatía tiroidea Síndrome de Kearns-Sayre Patología de nervio craneal Fatiga generalizada Enfermedad de motoneurona Síndrome de Lambert-Eaton Botulismo Miastenia congénita Miastenia por medicamentos

25
TRATAMIENTO Son 4 terapias básicas las que se pueden seguir.
Tratamiento sintomático con agentes anticolinesterasa. Tratamiento crónico inmunomodulador(glucocorticoides o inmunosupresores) Tratamiento rápido con imunomoduladores (plasmaféresis e inmunoglobulina intravenosa) Tratamiento quirúrgico: tiroidectomía Neurology. 2003;61(12):1652.
 Tratamiento rápido con imunomoduladores (plasmaféresis e inmunoglobulina intravenosa) Tratamiento quirúrgico: tiroidectomía. Neurology. 2003;61(12):1652.")
26
TRATAMIENTO Terapia sintomática Tiempo de inicio del efecto
Tiempo de máximo efecto Piridostigmina 10-15 minutos 2 horas Inmunoterapia crónica Prednisona 2-3 semanas 5-6 meses Azatioprina 6-12 meses 1-2 años Micofenolato de mofetilo 4-12 meses 1 año Ciclosporina 4-6 meses 8-12 meses Inmunoterapia rápida Plasmaferesis 1-7 días 1-3 semanas Inmunoglobulina intravenosa 1-2 semanas Cirugía 1-10 años Neurology. 2003;61(12):1652.
:1652.")
27
TRATAMIENTO Aproximación terapéutica: Individualización !!
Tratamiento depende: edad, severidad, presencia de sintomatología de involucro bulbar o respiratorio, progresión. Miastenia generalizada leve/moderada: piridostigmina. Severa o rápidamente progresiva: inmunoterapia rápida + inmunoterapia crónica. Semin Neurol. 2004;24(1):49.
:49.")
28
TRATAMIENTO Timectomía: No hay edad preferente
Se prefiere tener controlado al paciente para evitar riesgo perioperatorio. No siempre se puede extraer todo el tejido, por ello no se contempla como primera línea de tratamiento. Semin Neurol. 2004;24(1):49.
:49.")
29
TRATAMIENTO Primera línea: Piridostigminia : 30 mg 3-4 veces al día.
Hasta 120 mg c/6 horas Cuando no hay buena respuesta a dosis tope y/o efectos adversos se prefiere inmunoterapia. Efectos adversos: dolor abdominal y diarrea. Fasciculaciones y calambres. Neurology. 2003;61(12):1652.
:1652.")
30
TRATAMIENTO Segunda línea: inmunomoduladores
Remisión en 30% pacientes, mejoría en hasta 50% pacientes 1 mg/kg/día de prednisona. Se inician con 20 mg diarios y se incrementa 5 mgs cada 3-5 días hasta completar Azatioprina + glucocorticoides. Mejor respuesta. Se inicia a razón de 50 mg diarios por 2-3 semanas hasta llegar a 2-3 mg /kg. Neurology. 2003;61(12):1652.
:1652.")
31
TRATAMIENTO Inmunoterapia rápida, indicada en: Crisis miasténica
Preoperatorio Como puente para inicio de otra terapia Periódicamente como mantenimiento cuando otras terapias no responden bien. Semin Neurol. 2004;24(1):49.
:49.")
32
CRISIS MIASTÉNICA Condición que pone en riesgo la vida, que se define como debilidad con tal severidad de comprometer músculos respiratorios llevando a falla respiratoria con inmediata intubación. Factores precipitantes: infecciones, cirugías, embarazo, parto, medicamentos. J Clin Neuromuscul Dis. 2002;4(1):40.
:40.")
33
CRISIS MIASTÉNICA Se debe intubar y empezar inmunoterapia rápida.
Signos de alerta: Disnea, sobre todo en supino Disfagia severa Hipofonía, taquipnea, uso de músculos accesorios Disminución de Capacidad vital: - 30 ml/kg, aunque el paciente no tenga dificultad respiratoria. J Clin Neuromuscul Dis. 2002;4(1):40.
:40.")
34
MIASTENIA INDUCIDA POR MEDICAMENTOS
Los medicamentos pueden exacerbar o inducir la expresión de miastenia grave. Es una lista grande, sin embargo hay ciertos medicamentos mejor estudiados. La penicilamina induce miastenia grave en hasta el 1% de los consumidores. Ann Neurol. 1986;20(6):740.
:740.")
35
Se presentan meses después de iniciado el medicamento
Manifestaciones clínicas pueden ser con involucro ocular y/ generalizado debido a la producción de autoanticuerpos contra el receptor de acetilcolina. Se presentan meses después de iniciado el medicamento Descontinuando el medicamento los signos desaparecen en un lapso de 3-12 meses Arch Intern Med. 1997;157(4):399.
:399.")
36
Dentro de los medicamentos que deben ser evitados:
Aminoglucósidos Lidocaína Estatinas Arch Intern Med. 1997;157(4):399.
:399.")
37
Altas dosis de prednisona Sobredosis de acetilcolinesterasa
Medicamentos que tienen impacto negativo en pacientes con diagnostico d eMG: Altas dosis de prednisona Sobredosis de acetilcolinesterasa Anestésicos y relajantes musculares perioperatorios Arch Intern Med. 1997;157(4):399.
:399.")
38
SÍNDROME MIASTÉNICO: EATON LAMBERT
Desorden poco frecuente No hay estudios que nos permitan evaluar su real incidencia y prevalencia, sin embargo se ha podido determinar que se su incidencia llega a ser 1/ 2 millones. Es importante su diagnostico temprano debido a su alta asociación con cáncer pulmonar de células pequeñas ( hasta 50%)
")
39
Presentación como paraneoplásico o enfermedad autoinmune.
Existe producción de anticuerpos contra canales de calcio de la fibra presináptica. Presentación como paraneoplásico o enfermedad autoinmune. Clínicamente: Edad de presentación * Dificultad para iniciar la marcha o levantarse Fatiga muscular y calambres El compromiso muscular es simétrico Disfunción autonómica: xerostomía Ptosis Lancet. 1999;353(9147):117.
:117.")
40
A diferencia de miastenia grave:
Manifestación inicial con compromiso en extremidades Vs. Ocular Infrecuente compromiso orofaringeo Disminución en reflejos osteotendinosos Neurol Neurosurg Psychiatry. 2002;73(6):766
:766.")
41
El diagnóstico se confirma con la presencia de anticuerpos.
El tratamiento médico es seguido como en miastenia gravis, el fin es permitir mayor concentración de ACT en espacio sináptico. Se debe de buscar intencionalmente neoplasia pulmonar y de ovario. Lancet. 1999;353(9147):117.
:117.")
42
Lancet. 1999;353(9147):117.
:117.")
43
BOTULISMO Síndrome neuroparalítico
Resulta de la acción de la neurotoxína de la bacteria Clostridium botulinum. Actúa bloqueando la transmisión colinérgica impidiendo la transmisión de acetilcolina, afectando sinapsis muscarínicas y nicotínicas Am Fam Physician 2002 Apr 1;66(7):1388
:1388.")
44
Produce un total de 8 toxinas, la A, B, E causan daño al humano.
La forma de afección es por consumo de alimentos contaminados. La toxina se distribuye vía vascular y se une a un receptor específico en la fibra presiniaptica de las sinapsis de los ganglios y uniones neuromusculares.

45
La dosis letal mínima es de .0003 mcg/kg
Al penetrar a la célula produce unión disrupción en la estimulación y producción de acetilcolina. La dosis letal mínima es de mcg/kg Am Fam Physician 2002 Apr 1;66(7):1388
:1388.")
46
Dolor abdominal, diarrea.
Clínicamente: Inicio súbito Ausencia fiebre Dolor abdominal, diarrea. Déficit neurológico simétrico: nervios craneales + debilidad descendente Parálisis flácida de progresión descendente con dificultad respiratoria y disfunción autonómica Tendencia a bradicardia, normotensión Sensibilidad conservada Visión borrosa por midriasis arrefléctica Am Fam Physician 2002 Apr 1;66(7):1388
:1388.")
47
Definitivo: serología de la toxina.
Diagnóstico: pensar en botulismo, historia clínica completa y exploración detallada. Definitivo: serología de la toxina. Tratamiento: UTI, monitorización continua, contemplar intubación. Antitoxina: inmunoglobulina, intravenosa, aplicar lo mas pronto posible. Rev Infect Dis. 1984;6 Suppl 1:S202.

48
Es mortal si no se decta antes de la apricion de compromiso respiratorio.
La recuperación comienza en semanas y meses ( hasta 6), ya que se requiere de crecimiento de nuevos axones. Am Fam Physician 2002 Apr 1;66(7):1388
, ya que se requiere de crecimiento de nuevos axones. Am Fam Physician 2002 Apr 1;66(7):1388.")
Presentaciones similares