Descargar la presentación
La descarga está en progreso. Por favor, espere

2
Universidad Autónoma de Tamaulipas
Egresado de la facultad de Medicina Grado: Medico general Post Grado :Medico especialista en Psiquiatría

4
INSTITUTO DE ESTUDIOS SUPERIOES DE TAMAULIPAS
Profesor de grado y maestría.

5
Secretaria de salud del Estado de Veracruz
Modulo de Salud Mental Hospital Civil de Panuco Jefe del Área de Salud Mental

6
Secretaria de salud del Estado de Veracruz
Hospital Civil de Panuco & Jurisdiccion Sanitaria #1 del estado de Veracruz Profesor adjunto del Servicio de Educación Medica continua. Área de educación de pregrado.

7
Universitat Oberta de Catalanuya
Post Grado : Especialista en Conflictologia

8
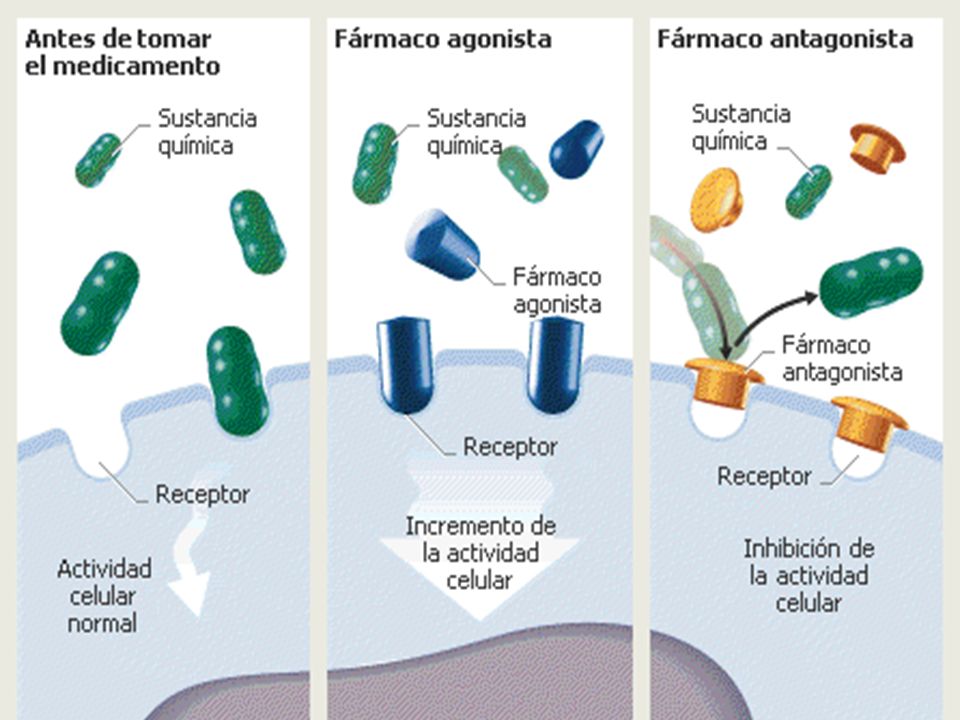
FARMACODINAMIA “Es lo que el Fármaco le hace al cuerpo”
Estudio de los efectos fisiológicos y bioquímicos de los fármacos y los mecanismos por los cuales se producen. El efecto farmacológico es el resultado de la interacción química o física entre el fármaco y la célula blanco. Los efectos pueden ser deseados (terapéuticos) o no deseados (RAMs). Existe una relación dosis-respuesta para cada tipo de efecto. “Es lo que el Fármaco le hace al cuerpo” 8
 o no deseados (RAMs). Existe una relación dosis-respuesta para cada tipo de efecto. Es lo que el Fármaco le hace al cuerpo 8.")
9
FARMACODINAMIA 9

10
RECEPTOR Componente de una célula u organismo que interactúa con un fármaco e inicia una cadena de fenómenos bioquímicos que originan los efectos observados del fármaco. Los receptores se han transformado en el foco de investigación de la farmacodinamia. El concepto de receptor se emplea en endocrinología, inmunología y biología molecular, es fundamental para explicar muchos aspectos de la regulación biológica. Pero además tiene implicaciones prácticas para el desarrollo de nuevos fármacos y para la toma de desiciones terapéuticas en la práctica clínica. Han sido aislados y caracterizados como macromoléculas, lo que ha permitido comprender las bases moleculares de la actividad de los fármacos. Los fármacos también se pueden unir a sitios no específicos (Tej. Adiposo) 10
 10.")
11
Clasificación de los fármacos según su mecanismo de acción
INSPECÍFICOS La acción biológica no depende de la estructura química, sino que sus fisicoquímicas (solubilidad, pKa, poder óxido reductor, etc.) Actúan en dosis relativamente altas. Fármacos con estructuras químicas muy variadas provocan reacciones biológicas semejantes. Pequeñas variaciones en sus estructuras no provocan alteraciones importantes de su acción biológica. Ejemplos: Agentes osmóticos (Manitol), Carbón activado.
 Actúan en dosis relativamente altas. Fármacos con estructuras químicas muy variadas provocan reacciones biológicas semejantes. Pequeñas variaciones en sus estructuras no provocan alteraciones importantes de su acción biológica. Ejemplos: Agentes osmóticos (Manitol), Carbón activado.")
12
ESPECÍFICOS: La acción biológica depende de la estructura química.
Actúan en dosis relativamente bajas. Presentan características estructurales comunes y la estructura fundamental está presente en todos ellos. Pequeñas variaciones en sus estructuras pueden provocan alteraciones importantes de su acción biológica, llevando a compuestos análogos o antagonistas. Poseen especificidad biológica, ejercen mayor efecto en un tejido que en otros. Ejemplos: casi todos.

13
ESPECÍFICOS: No mediados por receptores:
Drogas que actúan modificando el pH Agentes oxidantes o reductores Precipitadores de proteínas Agentes quelantes Mediados por receptores Proteínas reguladoras Proteínas de transporte Proteínas estructurales Acopladas a canales iónicos Enzimas 13

14
Enlaces Químicos que contribuyen a formar el complejo D-R
Fuerza de enlace Iónico Kcal/mol Van der Waals ,5 Kcal/mol Puentes de Hidrógeno Kcal/mol Covalente Kcal/mol 14

15
Importancia clínica del estudio de RECEPTORES
Determinan en gran parte las relaciones cuantitativas entre la dosis o concentración de un fármaco y sus efectos farmacológicos AFINIDAD y # TOTAL DE RECEPTORES La selectividad de la acción de los fármacos depende de los receptores Sirven de intermediarios en las acciones de los antagonistas farmacológicos Katzung p 13 15

16
Estudio de RECEPTORES Ensayos biológicos
Relación estructura química – actividad Agonistas Antagonistas Unión D-R Aislamiento de Receptores Secuenciación de AA en proteínas Aislamiento de genes y clonación Para próxima clase leer ¿Cómo se descubren los nuevos receptores? Y decidir si hacer lamina. 16

17
Aspectos de la actividad de los receptores de fármacos
Determinantes de la relación cuantitativa entre concentración del fármaco – respuesta farmacológica Su actividad como proteínas reguladoras y componentes de mecanismos de señalización Son elementos claves de los efectos terapéuticos y tóxicos de los fármacos Katzum p.15 17

18
1. Determinantes de la relación cuantitativa entre concentración del fármaco – respuesta farmacológica Interacción fármaco-receptor Curvas de concentración-efecto FARMACO Unión al receptor EFECTO Estudios de unión de radioligandos 18

19
Evolución histórica del concepto de receptor
John Langley (1905): Nicotina y curare Sustancia Receptiva Paul Ehrlich (1910): Especificidad antiparasitarios Quimioreceptores “Los fármacos no actúan si no se fijan” Toxicidad selectiva Índice Terapéutico: Dtoxica/Dterapéutica
: Nicotina y curare. Sustancia Receptiva. Paul Ehrlich (1910): Especificidad antiparasitarios. Quimioreceptores. Los fármacos no actúan si no se fijan Toxicidad selectiva. Índice Terapéutico: Dtoxica/Dterapéutica.")
20
Alfred Joseph Clark 1920: Teoría de la ocupación,
Proporcionalidad ocupación-efecto. La interacción requiere: afinidad del receptor por el fármaco y especificidad química. La intensidad del efecto farmacológico es función de la fracción de receptores ocupada por el fármaco. La fracción de receptores ocupada por el fármaco depende de la concentración de fármaco y del número total de receptores. El efecto máximo ocurre cuando todos los receptores se encuentran ocupados.

21
Teoría de Ocupación de Receptores
D + R DR Efecto K1 K2 K3 V1= k1 [D] [R] V2= k2 [DR] En equilibrio k1 [D] [R] = k2 [DR] K2 [D] [R] K [DR] = Kd Clark (1926) hace el primer tratamiento cuantitativo de la interacción D_R que sirve como base a la teoría de ocupación de receptores. Asume que: La magnitud del efecto es proporcional al No. de receptores ocupados La respuesta max. se obtiene cuando se ocupan todos los R. La relación D-R es 1:1 y es reversible. Con estas características de la unión D-R se puede aplicar la ley de acción de masas (ver diapo). (Ver apuntes carpeta azul) Notas: Kd= constante de disociación del complejo D-R .Es la concentración a la cual se ocupa el 50% de los R … Es una medida de la afinidad del agonista por el receptor. La Kd es constante para cada droga Kd= 1/afinidad Si ... [R] = [RT] - [DR] [Emax] C CE50 + C = [E] [RT] [D] Kd + [D] = [DR] 21
 hace el primer tratamiento cuantitativo de la interacción D_R que sirve como base a la teoría de ocupación de receptores. Asume que: La magnitud del efecto es proporcional al No. de receptores ocupados. La respuesta max. se obtiene cuando se ocupan todos los R. La relación D-R es 1:1 y es reversible. Con estas características de la unión D-R se puede aplicar la ley de acción de masas (ver diapo). (Ver apuntes carpeta azul) Notas: Kd= constante de disociación del complejo D-R .Es la concentración a la cual se ocupa el 50% de los R … Es una medida de la afinidad del agonista por el receptor. La Kd es constante para cada droga. Kd= 1/afinidad. Si ... [R] = [RT] - [DR] [Emax] C. CE50 + C. = [E] [RT] [D] Kd + [D] = [DR] 21.")
23
Afinidad Actividad intrinseca D + R DR Efecto Emax Efecto (%) [Emax] C
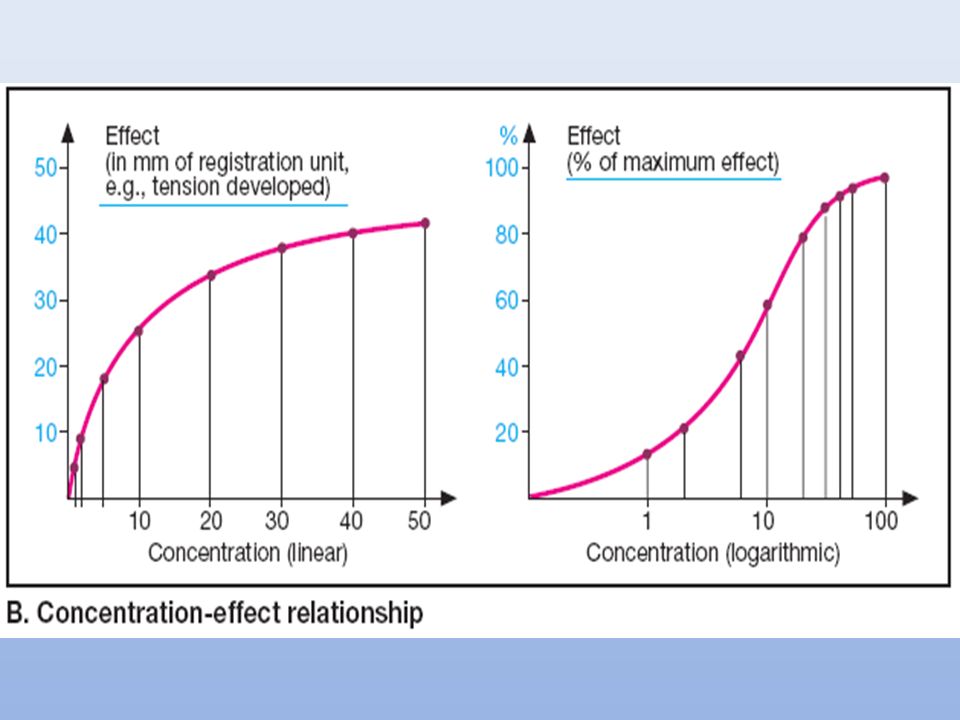
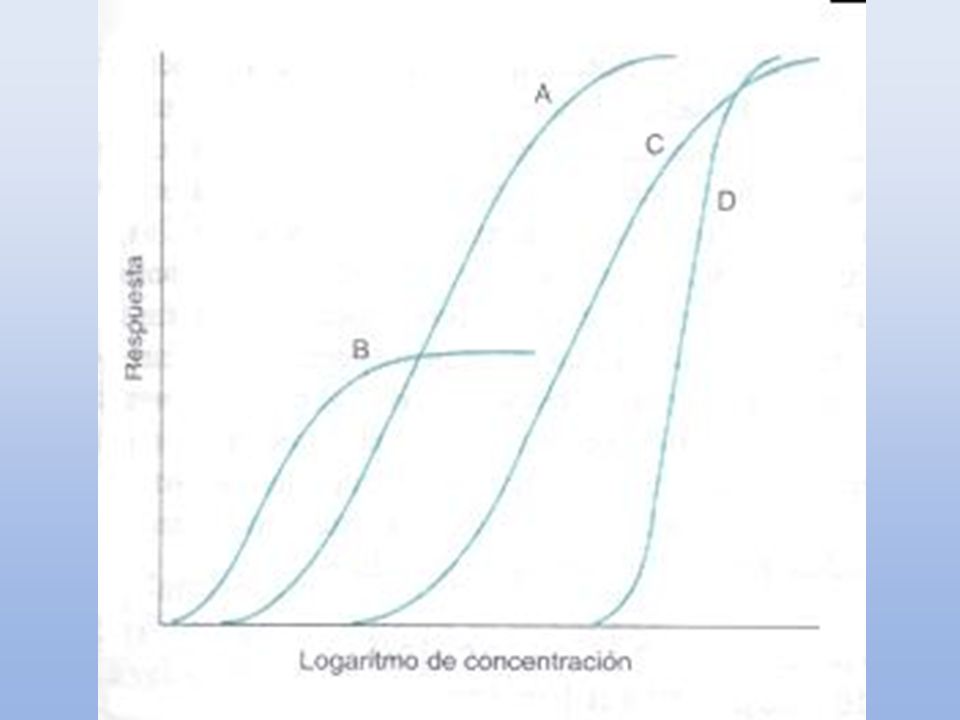
K1 K2 K3 D + R DR Efecto Emax CE50 Efecto (%) 1000 10 [D] = C 100 [Emax] C CE50 + C = [E] En un sistema ideal o in vitro, la relación entre la concetración del fármaco y su efecto, se describe mediante una curva hiperbólica, determinada por la ecuación de la DP, donde Emax: es el efectomáximo. C: es la concentración del fármaco CE50: es la concentración a la cual se produce la mitad del efecto máximo. La representación gráfica de los datosmejora graficando el efecto (ordenadas) vs el log. de la dosis (abcisas). La hiperbola se transforma en una sigmoide con una porción media lineal que facilita: la comparación gráfica de diferentes curvas expande la escala del eje de la concentracion a valores bajos y la comprime a valores altos donde ya los cambios son mínimos.(entre el 20 y el 80% de la respuesta queda en la parte lineal) Se puede graficar un mayor rango de dosis Se observa mejor la respuesta máxima Se puede también linearizar mediante la graficación de los inversos … Con el desarrollo de los radioligandos (agonistas o antagonistas) se pudo confirmar esta suposición de ocupación para diversos sistemas fármaco-receptor. Afinidad Actividad intrinseca 23
![Afinidad Actividad intrinseca D + R DR Efecto Emax Efecto (%) [Emax] C](http://slideplayer.es/slide/121575/1/images/23/Afinidad+Actividad+intrinseca+D+%2B+R+DR+Efecto+Emax+Efecto+%28%25%29+%5BEmax%5D+C.jpg "K1. K2. K3. D + R DR Efecto. Emax. CE50. Efecto (%) [D] = C [Emax] C. CE50 + C. = [E] En un sistema ideal o in vitro, la relación entre la concetración del fármaco y su efecto, se describe mediante una curva hiperbólica, determinada por la ecuación de la DP, donde. Emax: es el efectomáximo. C: es la concentración del fármaco. CE50: es la concentración a la cual se produce la mitad del efecto máximo. La representación gráfica de los datosmejora graficando el efecto (ordenadas) vs el log. de la dosis (abcisas). La hiperbola se transforma en una sigmoide con una porción media lineal que facilita: la comparación gráfica de diferentes curvas. expande la escala del eje de la concentracion a valores bajos y la comprime a valores altos donde ya los cambios son mínimos.(entre el 20 y el 80% de la respuesta queda en la parte lineal) Se puede graficar un mayor rango de dosis. Se observa mejor la respuesta máxima. Se puede también linearizar mediante la graficación de los inversos … Con el desarrollo de los radioligandos (agonistas o antagonistas) se pudo confirmar esta suposición de ocupación para diversos sistemas fármaco-receptor. Afinidad. Actividad intrinseca. 23.")
24
D + R DR Efecto Emax Efecto (%) CE50 [Emax] C [E] CE50 + C =
Semilogaritmica Efecto (%) 1 1000 Log [D] 100 10 CE50 K1 K2 K3 D + R DR Efecto Emax CE50 Efecto (%) 1000 10 [D] = C 100 Lineaveawer-Burk (inversos) 1000 10 1/E 100 Kd/Emax 1/Emax -1/Kd 1/[D] Kd E Emax.C Emax = En un sistema ideal o in vitro, la relación entre la concetración del fármaco y su efecto, se describe mediante una curva hiperbólica, determinada por la ecuación de la DP, donde Emax: es el efectomáximo. C: es la concentración del fármaco CE50: es la concentración a la cual se produce la mitad del efecto máximo. La representación gráfica de los datosmejora graficando el efecto (ordenadas) vs el log. de la dosis (abcisas). La hiperbola se transforma en una sigmoide con una porción media lineal que facilita: la comparación gráfica de diferentes curvas expande la escala del eje de la concentracion a valores bajos y la comprime a valores altos donde ya los cambios son mínimos.(entre el 20 y el 80% de la respuesta queda en la parte lineal) Se puede graficar un mayor rango de dosis Se observa mejor la respuesta máxima Se puede también linearizar mediante la graficación de los inversos … Con el desarrollo de los radioligandos (agonistas o antagonistas) se pudo confirmar esta suposición de ocupación para diversos sistemas fármaco-receptor. [Emax] C CE50 + C = [E] 24
![D + R DR Efecto Emax Efecto (%) CE50 [Emax] C [E] CE50 + C =](http://slideplayer.es/slide/121575/1/images/24/D+%2B+R+DR+Efecto+Emax+Efecto+%28%25%29+CE50+%5BEmax%5D+C+%5BE%5D+CE50+%2B+C+%3D.jpg "Semilogaritmica. Efecto (%) Log [D] CE50. K1. K2. K3. D + R DR Efecto. Emax. CE50. Efecto (%) [D] = C Lineaveawer-Burk (inversos) /E Kd/Emax. 1/Emax. -1/Kd. 1/[D] 1 Kd + 1. E Emax.C Emax. = En un sistema ideal o in vitro, la relación entre la concetración del fármaco y su efecto, se describe mediante una curva hiperbólica, determinada por la ecuación de la DP, donde. Emax: es el efectomáximo. C: es la concentración del fármaco. CE50: es la concentración a la cual se produce la mitad del efecto máximo. La representación gráfica de los datosmejora graficando el efecto (ordenadas) vs el log. de la dosis (abcisas). La hiperbola se transforma en una sigmoide con una porción media lineal que facilita: la comparación gráfica de diferentes curvas. expande la escala del eje de la concentracion a valores bajos y la comprime a valores altos donde ya los cambios son mínimos.(entre el 20 y el 80% de la respuesta queda en la parte lineal) Se puede graficar un mayor rango de dosis. Se observa mejor la respuesta máxima. Se puede también linearizar mediante la graficación de los inversos … Con el desarrollo de los radioligandos (agonistas o antagonistas) se pudo confirmar esta suposición de ocupación para diversos sistemas fármaco-receptor. [Emax] C. CE50 + C. = [E] 24.")
25
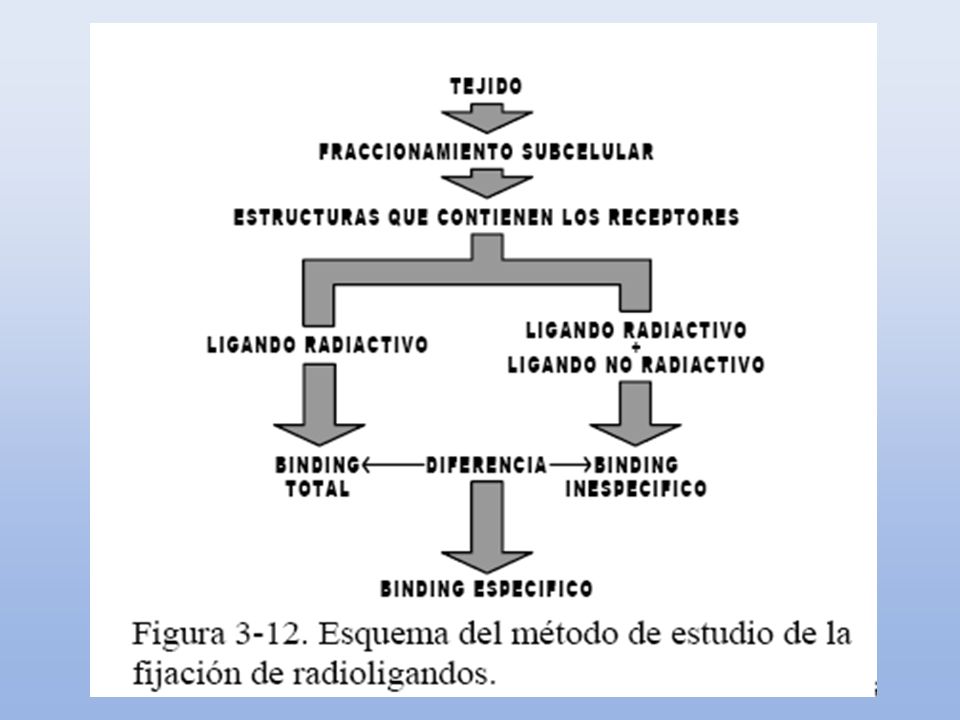
ESTUDIOS DE UNIÓN CON RADIOLIGANDOS
Incubación de un tejido con concentraciones crecientes de fármaco con alta afinidad y especificidad para un receptor, marcado radiactivamente. Capacidad de cuantificar la formación del complejo droga – receptor. Medición de la afinidad real de una droga (KD) por su receptor. Se llaman también estudios de saturaci´pn. Necesitamos: Disponer de una droga marcada radioactivamente, que se una al receptor con: a) Alata afinidad, b) alto grado de selectividad. 25
 por su receptor. Se llaman también estudios de saturaci´pn. Necesitamos: Disponer de una droga marcada radioactivamente, que se una al receptor con: a) Alata afinidad, b) alto grado de selectividad. 25.")
27
ESTUDIOS DE UNIÓN CON RADIOLIGANDOS
27

28
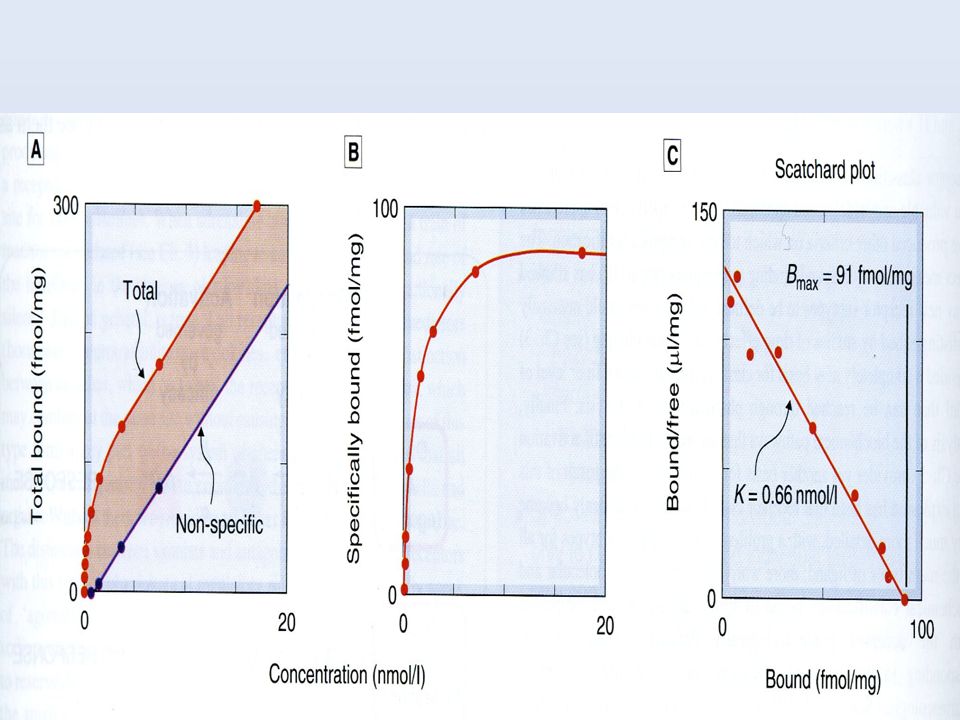
ESTUDIOS DE UNIÓN CON RADIOLIGANDOS
Kd Bmax

29
ESTUDIOS DE UNIÓN CON RADIOLIGANDOS
K3

31
Características de la unión D-R
Medible Dependiente de la concentración y de la afinidad del fármaco por el receptor Saturable Específica Reversible

32
// // // // Kd D + R DR Efecto CE50 CE50 Kd Actividad intrinseca
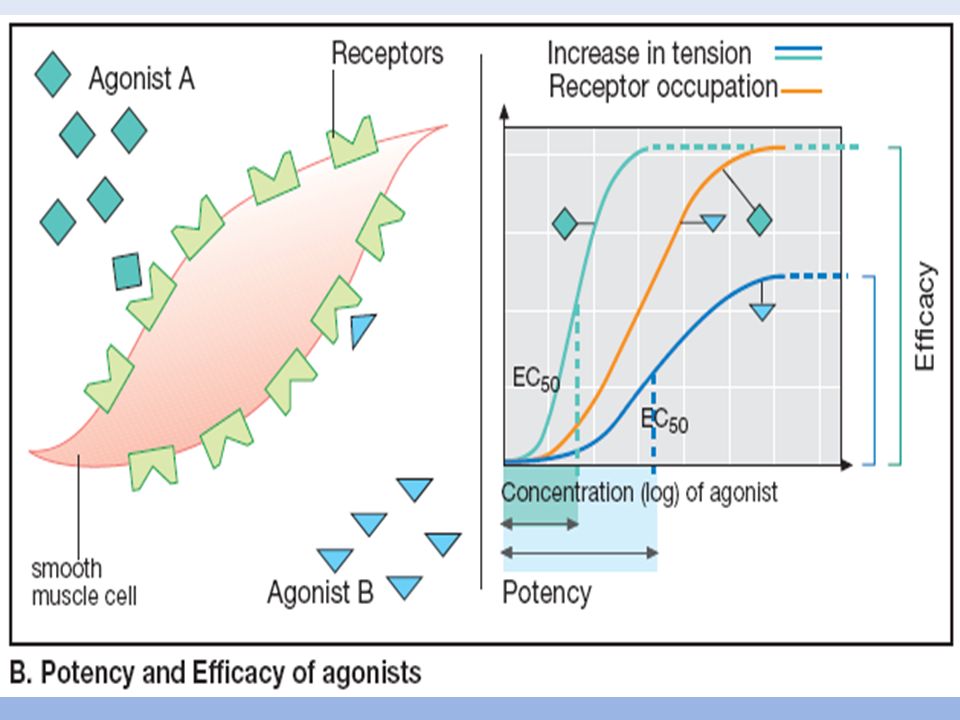
K3 o CE50 Efecto (%) (E) 10 Conc. Fármaco (C) 100 // Kd Fármaco unido al Receptor (B) 10 Conc. Fármaco (C) 100 // // // E= Emax . C CE50 + C B= Bmax . C Kd + C CE Kd Actividad intrinseca Potencia Afinidad Eficacia
 (E) 10. Conc. Fármaco (C) 100. // Kd. Fármaco unido al Receptor (B) 10. Conc. Fármaco (C) 100. // // // E= Emax . C. CE50 + C. B= Bmax . C. Kd + C. CE50 Kd Actividad intrinseca. Potencia Afinidad Eficacia.")
33
Ariens (1954): actividad intrinseca y Stephenson (1956): agonista parcial y RR
La interacción F-R comprende dos fases: Formación del complejo F-R y producción del efecto. Además de afinidad, el fármaco debe tener capacidad para producir el efecto biológico (actividad intrínseca). La actividad intrínseca tiene valores entre 1 y 0. Es igual a 1 para los agonistas completos, es igual a 0 para los antagonistas y posee valores entre 0 y 1 para los agonistas parciales.
. La actividad intrínseca tiene valores entre 1 y 0. Es igual a 1 para los agonistas completos, es igual a 0 para los antagonistas y posee valores entre 0 y 1 para los agonistas parciales.")
34
Clasificación de fármacos en relación con la actividad intrínseca
Agonistas: se unen al receptor y poseen actividad intrínseca. Antagonistas: se unen al receptor pero carecen de actividad intrínseca. Agonistas parciales: se unen al receptor y posen actividad intrínseca inferior a la presentada por los agonistas puros. Agonistas inversos: se unen al receptor y tienen actividad intrínseca con efectos opuestos a los del agonista puro.

37
D + R DR Efecto K1 K2 k3 o Los receptores existen en dos estados activo (Ra) e inactivo (Ri) (ej. Abierto y cerrado del canal iónico, activo e inactivo de la proteina cinasa o las conformaciones que le permiten acoplarse o no a la proteína G) Si ambos estados están en equilibrio y en ausencia del fármaco predomina el estado inactivo, la señal que se genera es pequeña. En presencia de un fármaco el efecto va a depender de la afinidad relativa del fármaco por Ra o Ri. Si el fármaco presenta mayor afinidad por la configuración activa, arraatra el equilibrio hacia la conformación activa y se activará el receptor (Un agonista completo tiene suficiente afinidad por Ra para en condiciones de saturación desplazar completamente el equilibrio hacia en estado activo). Otro compuesto (que puede ser análogo en su estructura) se une al mismo receptor con afinidad por Ra>Ri producirá un efecto menor aún a concentraciones de saturación y se ha denominado agonista parcial. Nota: En sentido absoluto todos los agonistas serían parciales, ya que la selectividad por Ra o Ri no es absoluta. Un medicamento que se una con igual afinidad a ambos estados conformacionales, no modificará el equilibrio y actuará como un antagonista competitivo. Y si el medicamentotiene afinidad preferente por Ri, producirá un efecto contrario al agonista y se conocen como agonistas inversos. Estudios bioquímicos de mutación de receptores han permitido cambiar el equilibrio Ra/Ri que apoyan este modelo y existen modelos computarizados que pueden ser aplicados a datos experimentales. 37
 e inactivo (Ri) (ej. Abierto y cerrado del canal iónico, activo e inactivo de la proteina cinasa o las conformaciones que le permiten acoplarse o no a la proteína G) Si ambos estados están en equilibrio y en ausencia del fármaco predomina el estado inactivo, la señal que se genera es pequeña. En presencia de un fármaco el efecto va a depender de la afinidad relativa del fármaco por Ra o Ri. Si el fármaco presenta mayor afinidad por la configuración activa, arraatra el equilibrio hacia la conformación activa y se activará el receptor (Un agonista completo tiene suficiente afinidad por Ra para en condiciones de saturación desplazar completamente el equilibrio hacia en estado activo). Otro compuesto (que puede ser análogo en su estructura) se une al mismo receptor con afinidad por Ra>Ri producirá un efecto menor aún a concentraciones de saturación y se ha denominado agonista parcial. Nota: En sentido absoluto todos los agonistas serían parciales, ya que la selectividad por Ra o Ri no es absoluta. Un medicamento que se una con igual afinidad a ambos estados conformacionales, no modificará el equilibrio y actuará como un antagonista competitivo. Y si el medicamentotiene afinidad preferente por Ri, producirá un efecto contrario al agonista y se conocen como agonistas inversos. Estudios bioquímicos de mutación de receptores han permitido cambiar el equilibrio Ra/Ri que apoyan este modelo y existen modelos computarizados que pueden ser aplicados a datos experimentales. 37.")
38
INTEGRACIÓN FUNCIONALIDAD – OCUPACIÓN DEL RECEPTOR

39
INTEGRACIÓN FUNCIONALIDAD – OCUPACIÓN DEL RECEPTOR

40
Agonista parcial: tiene actividad intrínseca “per se” aunque inferior al agonista puro (menor eficacia). En presencia de agonista puro se comporta como antagonista competitivo.

41
Furchgot (1955): Receptores de reserva.
Un fármaco con una eficacia muy alta podría alcanzar el efecto máximo sin necesidad de ocupar todos los receptores. La fracción no ocupada se conoce como receptores de reserva. Su existencia se demuestra con bloqueadores irreversibles. Un fármaco de baja eficacia (agonista parcial), no es capaz de alcanzar el efecto máximo aunque ocupe todos los receptores
, no es capaz de alcanzar el efecto máximo aunque ocupe todos los receptores.")
43
Receptores no ocupados o de reserva
Acoplamiento fármaco-receptor

44
Interacción entre fármacos
Antagonismo Sinérgismo Fisiológico Químico Farmacológico Competitivo No competitivo Mixto Suma Potenciación 44

45
ANTAGONISMO Antagonismo fisiológico: Los fármacos tienen acciones opuestas y actúan a través de receptores distintos. Antagonismo químico: Los fármacos, en base a su naturaleza química, reaccionan entre sí lo que conduce a la inactivación del fármaco activo. Antagonismo farmacológico: Implica la unión a un mismo receptor: Competitivo, No competitivo

46
Antagonismo competitivo
El agonista y el antagonista compiten por el mismo lugar de unión al receptor (de carácter reversible al aumentar la dosis de agonista) La droga se une al mismo receptor que el agonista El efecto es superable, siempre se alcanza Emax (se puede lograr el 100% de ocupación) con dosis mayores de AGO no se modifica la efcacia La curva se desplaza a la derecha Aumenta CE50 dependiendo de la conc. del antagonista Necesito mayor concentración del agonista para obtener tanto la ocupación maxima como el efecto maximo. Esto clinicamente se traduce … 1.- Si se administra un antagonista competitivo de una sustanecia endógena: El grado de inhibición depende de la conc. del antagonista Ej del propranolol ... La magnitud y duración de su acción dependerá de su concentración en plasma ( sitio de acción), que es influenciadas por la depuración hepática (variable farmacocinética) ... Los resultados de una miama dosispuede producir efectos variables entre pacientes ... Ajuste de dosis que deben ser diferentes entre pacientes. La variabilidad de la respuesta clínica a un antagonista competitivo, depende de la conc. del agonista. El efecto bloqueante del propranolol sobre la frecuencia cardíaca es diferente en condiciones de reposo, que la disminución e FC posterior a ejercicio o estrés emocional, donde el efecto de incremento deliberación de NA puede contrarrestar el efecto. 2. Debe tenerse en cuenta cuando se administran varios fármacos conjuntamente: Ej. Un paciente asmático que recibe AGOB2 (salbutamol) broncodilatador, si además es hipertenso no podemos darle propanolol que es un antagonista B no selectivo, ya que interferiría con el efecto de la medicación que recibe para el asma. 46
 La droga se une al mismo receptor que el agonista. El efecto es superable, siempre se alcanza Emax (se puede lograr el 100% de ocupación) con dosis mayores de AGO no se modifica la efcacia. La curva se desplaza a la derecha. Aumenta CE50 dependiendo de la conc. del antagonista. Necesito mayor concentración del agonista para obtener tanto la ocupación maxima como el efecto maximo. Esto clinicamente se traduce … 1.- Si se administra un antagonista competitivo de una sustanecia endógena: El grado de inhibición depende de la conc. del antagonista. Ej del propranolol ... La magnitud y duración de su acción dependerá de su concentración en plasma ( sitio de acción), que es influenciadas por la depuración hepática (variable farmacocinética) ... Los resultados de una miama dosispuede producir efectos variables entre pacientes ... Ajuste de dosis que deben ser diferentes entre pacientes. La variabilidad de la respuesta clínica a un antagonista competitivo, depende de la conc. del agonista. El efecto bloqueante del propranolol sobre la frecuencia cardíaca es diferente en condiciones de reposo, que la disminución e FC posterior a ejercicio o estrés emocional, donde el efecto de incremento deliberación de NA puede contrarrestar el efecto. 2. Debe tenerse en cuenta cuando se administran varios fármacos conjuntamente: Ej. Un paciente asmático que recibe AGOB2 (salbutamol) broncodilatador, si además es hipertenso no podemos darle propanolol que es un antagonista B no selectivo, ya que interferiría con el efecto de la medicación que recibe para el asma. 46.")
47
Emax se alcanza, CE50 aumenta

48
48

49
Antagonismo no competitivo
Se une a otro lugar del receptor que impide que el agonista ejerza su efecto biológico (no reversible al aumentar la concentración del agonista) La recuperación de la respuesta va a depender de la síntesis de novo de la molécula efectora y no de la eliminación del fármaco circulante. Se forman enlaces covalentes. Ej organofosforados inhiben la Acetilcolinesterasa. Fenoxibenzamina para disminuir la PA en pacientes con feocromocitoma ... Se mantiene el bloqueo asi el tumor libere periodicamente cantidades considerables de catecolaminas. Riesgo ... Si hay un efecto excesivo del fármaco, no puede revertirse con agonistas alfa adrenérgicos, debe producirse un efecto vasopresor utilizando antagonistas fisiológicos. 49
 La recuperación de la respuesta va a depender de la síntesis de novo de la molécula efectora y no de la eliminación del fármaco circulante. Se forman enlaces covalentes. Ej organofosforados inhiben la Acetilcolinesterasa. Fenoxibenzamina para disminuir la PA en pacientes con feocromocitoma ... Se mantiene el bloqueo asi el tumor libere periodicamente cantidades considerables de catecolaminas. Riesgo ... Si hay un efecto excesivo del fármaco, no puede revertirse con agonistas alfa adrenérgicos, debe producirse un efecto vasopresor utilizando antagonistas fisiológicos. 49.")
50
Disminución de pendiente y Emax, CE50 no se modifica.

51
Competitivo No Competitivo
Modifica la afinidad Modifica la eficacia

52
52

53
SINERGISMO Interacción entre un agonista y un agonista parcial
SUMA 2 AGO, o un AGO y un AGO parcial … a concentraciones bajas. A altas concentraciones no se ve el efecto sumatorio porque los receptores estan ocupados. Importante: Para evaluar tanto sumación comopotenciación hay que utilizar dosis bajas, a menos que en los casos de potenciación se quiera ver si se incrementa la respuesta máxima. POTENCIACION AGO + agente sin efecto perse 2 AGO, o un AGO y un AGO parcial … a concentraciones bajas Ver anterior y próximas 53

54
Interacción entre un agonista y un agonista parcial
HA HA + IMP HA+IMP Interacción entre la histamina y el agonista parcial del receptor H2 impromidina, en tiras de miocardio aisladas de ventriculo humano. Izq. HA sola. Derecha impromidina sola () y en presencia de una concentración constante de HA. El otro grafico es la curva de HA sola, en presencia de una concentreación única de inpromidina. Receptor Pharmacology p..22 HA 54
 y en presencia de una concentración constante de HA. El otro grafico es la curva de HA sola, en presencia de una concentreación única de inpromidina. Receptor Pharmacology p..22. HA. 54.")
55
M 55

56
Potenciación del efecto de un fármaco:
Aumentando su concentración a nivel del receptor Aumentando su afinidad por el receptor Aumentando el número de receptores funcionantes Supersensibilización Desensibilización Modificando su actividad intrínseca (1)Ej La cocaína inhibe la recaptación de NA, incrementa su concentración a nivel del R e incrementa su efecto. Inhibidores de la acetilcolinesterasa incrementa el efecto de la ACh. (2) Es posible desde el punto de vista teórico (3) Desensibilización … Homóloga: Insulina Heteróloga: Hormona tiroidea incrementa el No. de R Beta en el músculo cardíaco taquicardia. Supersensibilización: Uso prolongado de antagonista cuidado cuando se suspende. Ej. Clonidina (4) Ej. AGO B2 … AMPc glucogenolisis. Si se administran inhibidores de la fosfodiesterasa, enzima que degrada al AMPc, se potenciaría el efecto. 56
Ej La cocaína inhibe la recaptación de NA, incrementa su concentración a nivel del R e incrementa su efecto. Inhibidores de la acetilcolinesterasa incrementa el efecto de la ACh. (2) Es posible desde el punto de vista teórico. (3) Desensibilización … Homóloga: Insulina. Heteróloga: Hormona tiroidea incrementa el No. de R. Beta en el músculo cardíaco taquicardia. Supersensibilización: Uso prolongado de antagonista cuidado cuando se. suspende. Ej. Clonidina. (4) Ej. AGO B2 … AMPc glucogenolisis. Si se administran inhibidores de la fosfodiesterasa, enzima que degrada al AMPc, se potenciaría el efecto. 56.")
57
Aspectos de la actividad de los receptores de fármacos
Determinantes de la relación cuantitativa entre concentración del fármaco – respuesta farmacológica Su actividad como proteínas reguladoras y componentes de mecanismos de señalización Son elementos claves de los efectos terapéuticos y tóxicos de los fármacos Katzum p.15 57

58
TIPOS DE RECEPTORES Canales Iónicos
Receptores para sustancias liposolubles Con actividad tirosina quinasa Receptores acoplados a proteínas G

59
1. CANAL IÓNICO

60
2. RECEPTOR PARA SUSTANCIAS LIPOSOLUBLES

61
intracelular de los glucocorticoides
Mecanismo de acción intracelular de los glucocorticoides

62
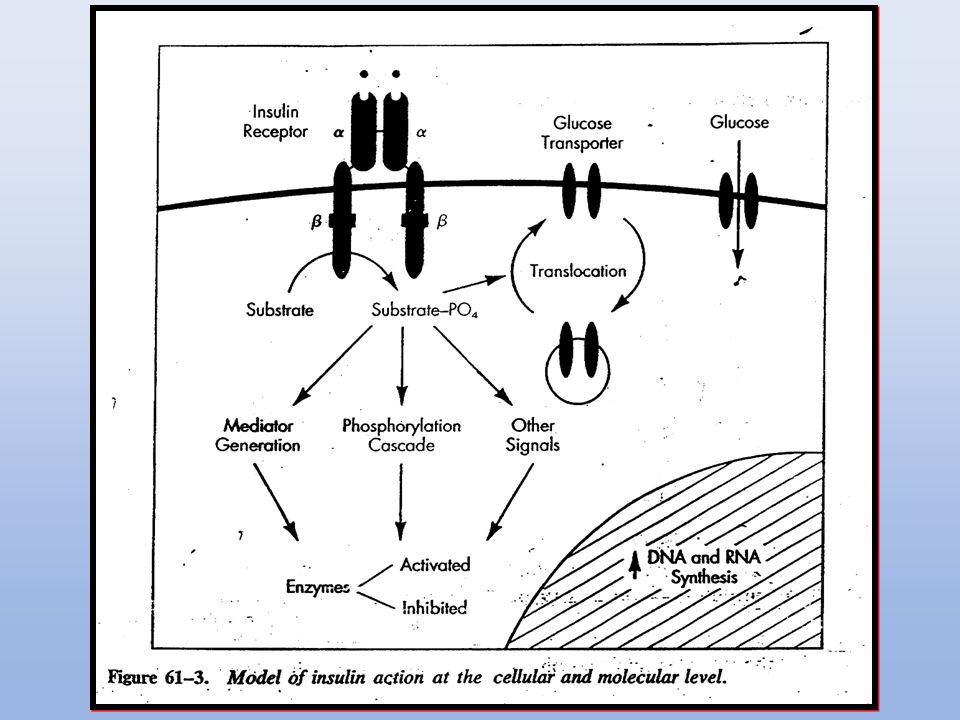
3. RECEPTORES CON ACTIVIDAD TIROSINA QUINASA

64
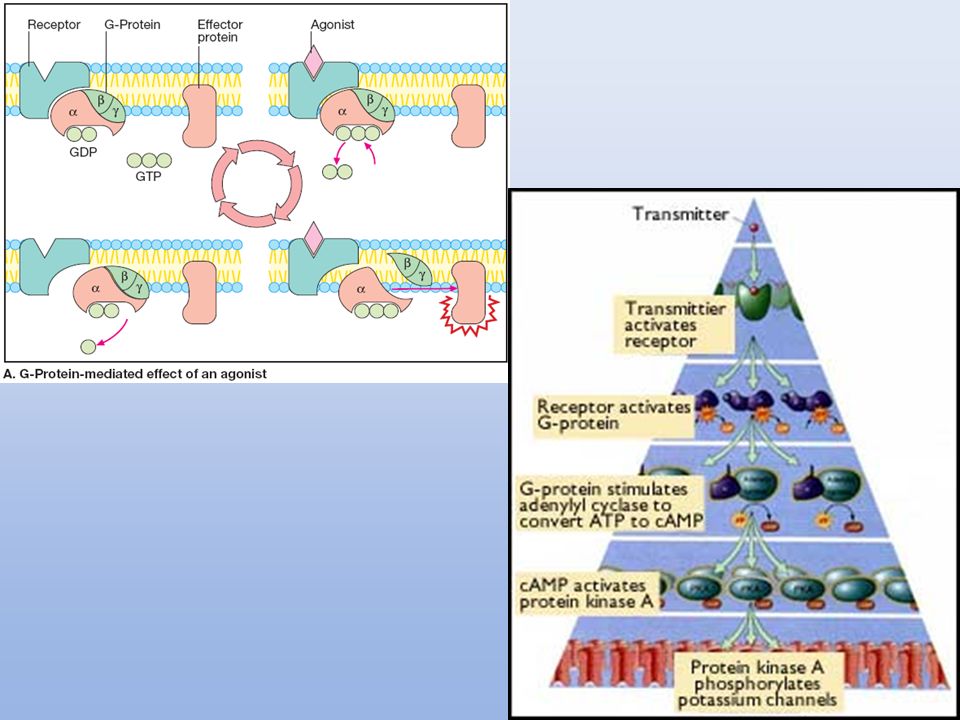
4. RECEPTORES ACOPLADOS A PROTEÍNAS G

65
Señalización mediada por AMPc

66
Señalización mediada por inositol trifosfato

68
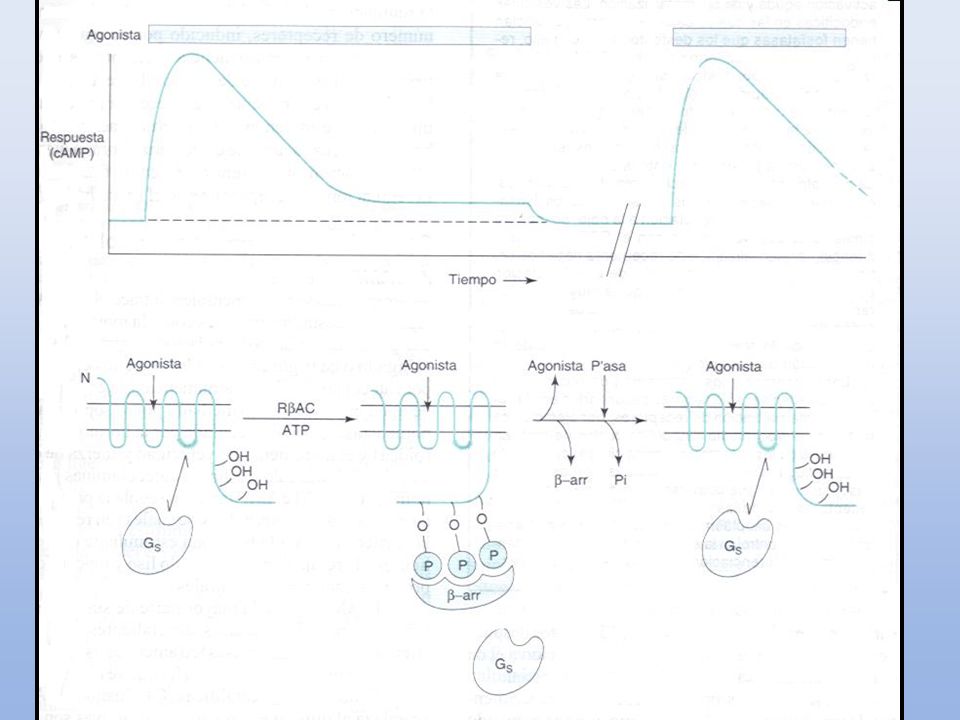
REGULACIÓN DE RECEPTORES
Desensibilización (taquifilaxia): Pérdida de respuesta a la acción de un ligando (fármaco) cuando éste se administra de manera continuada o repetida. Periodo de minutos/horas (Tolerancia: días/semanas) Regulación homóloga: el ligando afecta únicamente al receptor ocupado por el propio ligando Regulación heteróloga: el receptor se ve afectado por la unión de un ligando a otros receptores
: Pérdida de respuesta a la acción de un ligando (fármaco) cuando éste se administra de manera continuada o repetida. Periodo de minutos/horas (Tolerancia: días/semanas) Regulación homóloga: el ligando afecta únicamente al receptor ocupado por el propio ligando. Regulación heteróloga: el receptor se ve afectado por la unión de un ligando a otros receptores.")
69
Mecanismos (homóloga):
a) Disminución en la afinidad por el ligando b) Inhibición del acoplamiento entre el receptor y los elementos de respuesta celular c) Reducción en el número de receptores - internalización (endocitosis) - ↑ degradación - ↓ disminución de la síntesis de nuevos receptores
 Disminución en la afinidad por el ligando. b) Inhibición del acoplamiento entre el receptor y los elementos de respuesta celular. c) Reducción en el número de receptores. - internalización (endocitosis) - ↑ degradación. - ↓ disminución de la síntesis de nuevos receptores.")
72
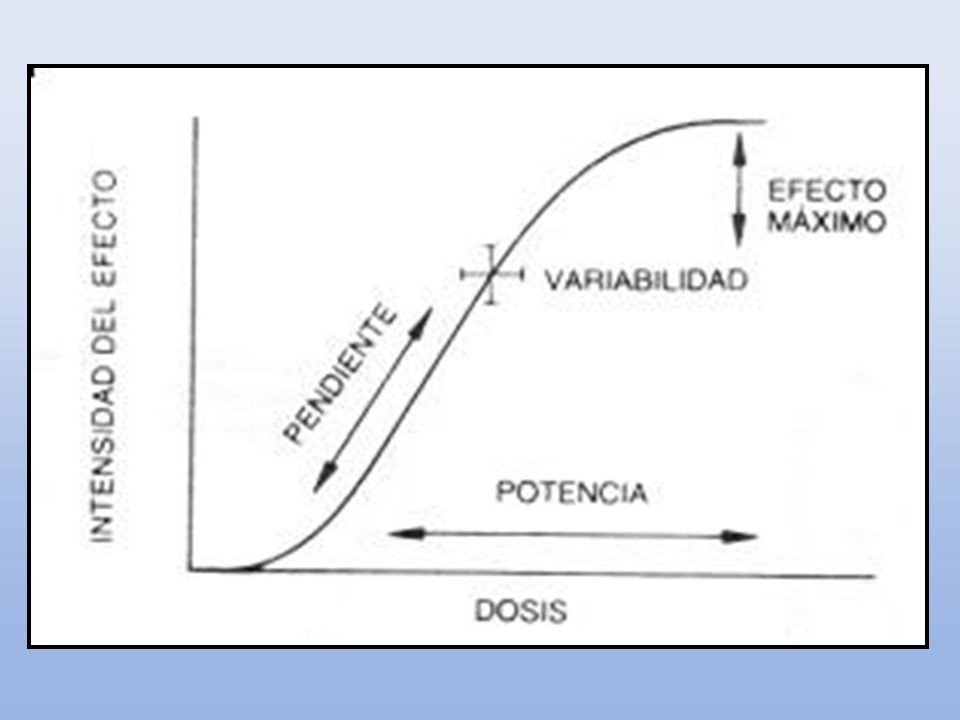
POTENCIA EFICACIA

75
Aspectos de la actividad de los receptores de fármacos
Determinantes de la relación cuantitativa entre concentración del fármaco – respuesta farmacológica Su actividad como proteínas reguladoras y componentes de mecanismos de señalización Son elementos claves de los efectos terapéuticos y tóxicos de los fármacos Katzum p.15 75

76
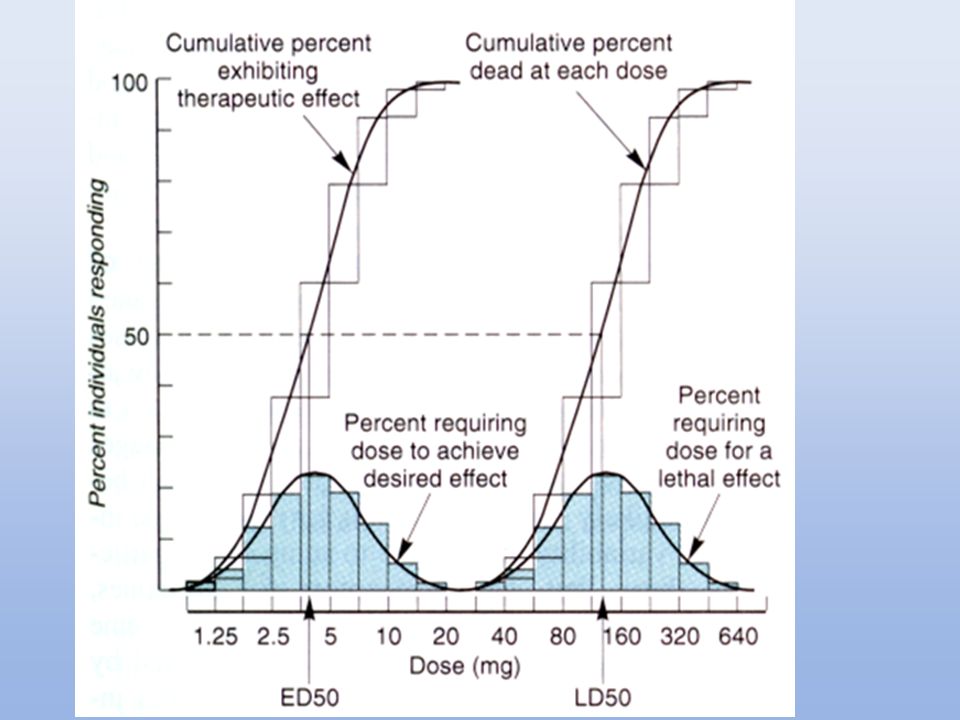
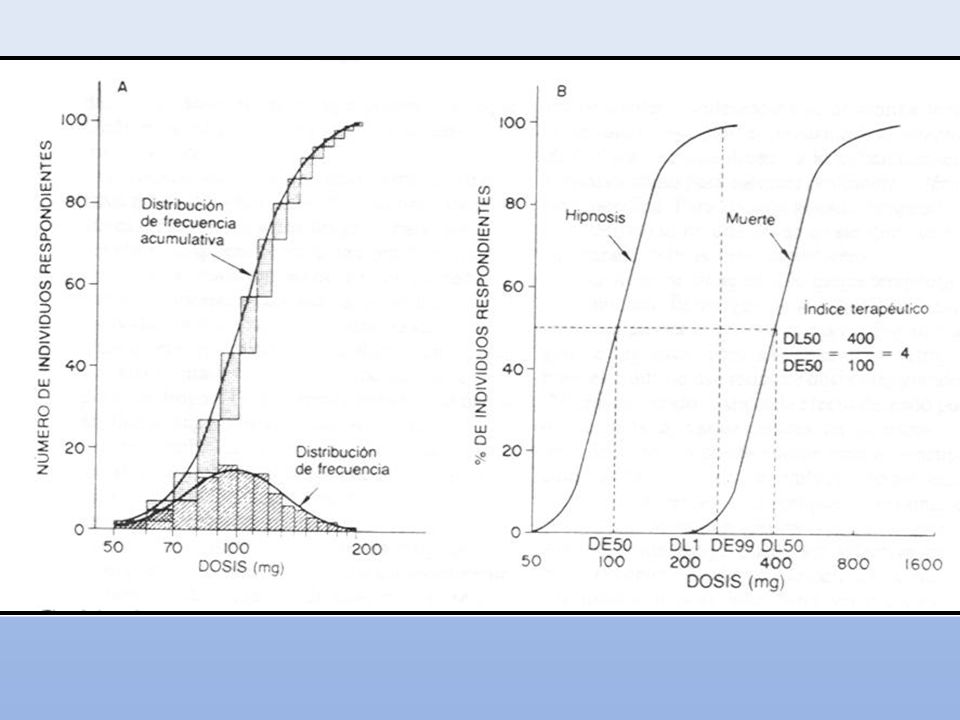
CURVAS CUANTALES Dosis efectiva cincuenta (DE50): dosis a la cual se produce un efecto específico en el 50% de los individuos. Dosis tóxica cincuenta (DT50): dosis requerida para producir un efecto tóxico determinado en el 50% de los individuos. Dosis Letal Cincuenta (DL50): dosis a la cual mueren el 50% de los animales de experimentación. Utilidad: Comparación de potencias entre drogas. Selectividad para producir un determinado efecto. Índice terapéutico (IT): DT50/DE50. Utilidad: margen de seguridad de una droga en particular para producir un efecto específico.
: dosis requerida para producir un efecto tóxico determinado en el 50% de los individuos. Dosis Letal Cincuenta (DL50): dosis a la cual mueren el 50% de los animales de experimentación. Utilidad: Comparación de potencias entre drogas. Selectividad para producir un determinado efecto. Índice terapéutico (IT): DT50/DE50. Utilidad: margen de seguridad de una droga en particular para producir un efecto específico.")
79
Relaciones entre efectos terapéuticos y tóxicos de un fármaco
Receptor Efector Respuesta Benéfica Tóxica 1. D + R DR X Benéfica Tóxica X 2. D + R DR Y R1 DR1 R2 DR2 Benéfica Tóxica X 3. D+ Y

80
GRACIAS

Presentaciones similares


















![1 KM 1 1 Vo= Vmax [S] + Vmax Y = m x + b = m = b.](/2/161459/big_thumb.jpg "1 KM 1 1 Vo= Vmax [S] + Vmax Y = m x + b = m = b.>")




