Descargar la presentación
La descarga está en progreso. Por favor, espere

1
MIOPATIAS Dr. Marco A. Reza Orozco
PROFESOR TITULAR: Dr. Enrique Díaz Greene PROFESOR ADJUNTO: Dr. Federico Rodríguez Weber SUPERVISÓ: Dra. Pamela Vazquez

2
DEFINICIÓN Goldman: Cecil Medicine, 23rd ed.
Enfermedad del musculo esquelético Alteración de la estructura primaria o alteración en la función del musculo. Goldman: Cecil Medicine, 23rd ed.

3
Músculo Músculo Estriado Músculo Esquelético Músculo Cardiaco Músculo Liso Músculo Visceral

4
Guyton: textbook of Medical Physiology, 11th ed.

5
Goldman: Cecil Medicine, 23rd ed.

6
Antecedentes heredofamiliares, edad inicio, evolución Exploración
Interrogatorio Antecedentes heredofamiliares, edad inicio, evolución Exploración Contracción, relajación y reposo muscular. Fatiga. Gabinete Enzimas musculares Electromiografía Biopsia muscular

7
Síntomas Negativos: Debilidad Proximal > distal Edad de presentación (década) Permanente vs intermitente Tiempo de evolución aguda (< 4 semanas) miopatías inflamatorias Subaguda ( 4 – 8 semanas) Crónica ( > 8 semanas) distrofias / congénitas Recaídas: monofásico vs polifásico Debilidad objetiva síntoma más confiable
 miopatías inflamatorias. Subaguda ( 4 – 8 semanas) Crónica ( > 8 semanas) distrofias / congénitas. Recaídas: monofásico vs polifásico. Debilidad objetiva síntoma más confiable.")
8
Síntomas positivos: Mialgia Episódicas (metabólicas) Constante (inflamatorias) No específico, es poco común Calambre muscular Generalmente benigno EMG: descargas rápidas

9
Síntomas positivos: Mialgia Episódicas (metabólicas) Constante (inflamatorias) No específico, es poco común Calambre muscular Generalmente benigno EMG: descargas rápidas

10
Contractura muscular Poco común Mayor duración que un calambre Paciente con deficiencia enzimática glagolítica EMG: silencio eléctrico Miotonía Ausencia de relajación muscular después de una contracción voluntaria Mejoran con el ejercicio repetido “calentamiento” Si empeoran con el ejercicio se denomina “paramiotonía” Empeoran con el frío Se debe a despolarizaciones repetitivas de la membrana muscular

11
1. Mm proximales de las extremidades
Es el más común Poco específico Puede involucrar a los mm flexores y extensores cervicales Goldman: Cecil Medicine, 23rd ed.

12
Goldman: Cecil Medicine, 23rd ed.
PATRONES DE DEBILDAD 2. Mm distales en las extremidades superiores (mm extensores) e inferiores (compartimento anterior y posterior) Más común se encuentra en neuropatías Ejemplos: distrofia miotónica, distrofia distal, titinopatía, zaspopatía y miositis por cuerpos de inclusión Goldman: Cecil Medicine, 23rd ed.
 e inferiores (compartimento anterior y posterior) Más común se encuentra en neuropatías. Ejemplos: distrofia miotónica, distrofia distal, titinopatía, zaspopatía y miositis por cuerpos de inclusión. Goldman: Cecil Medicine, 23rd ed.")
13
Goldman: Cecil Medicine, 23rd ed.
PATRONES DE DEBILDAD 3. Debilidad proximal superior de los músculos periescapulares y de las extremidades inferiores del compartimento anterior (escapulo-peroneal) Al asociarse con debilidad facial distrofia fascio-escapulo-humeral Se asocia: distrofia escapuloperoneal, distrofia de Emery-Dreifuss, deficiencia de Maltasa y algunas miopatías congénitas Goldman: Cecil Medicine, 23rd ed.
 Al asociarse con debilidad facial distrofia fascio-escapulo-humeral. Se asocia: distrofia escapuloperoneal, distrofia de Emery-Dreifuss, deficiencia de Maltasa y algunas miopatías congénitas. Goldman: Cecil Medicine, 23rd ed.")
14
PATRONES DE DEBILDAD 4. MsTs (distal) muñecas y dedos flexores; MsPs (proximal) cuadriceps Patognomónico de la miositis por cuerpos de inclusión La debilidad es asimétrica (poco común) Goldman: Cecil Medicine, 23rd ed.
 Goldman: Cecil Medicine, 23rd ed.")
15
Goldman: Cecil Medicine, 23rd ed.
PATRONES DE DEBILDAD 5. Mm óculo-faríngeos Ptosis + oftalmoplegia sin diplopia + debilidad faringea distrofia oculofaringea Ptosis + oftalmoplegia sin alteración faringea prominente miopatía mitocondrial Ptosis + debilidad facial sin oftalmoplegia o debildiad faringea distrofia miotónica Goldman: Cecil Medicine, 23rd ed.

16
“dropped head syndrome” Compartido por: ELA y MG
PATRONES DE DEBILDAD Mm extensores del cuello “dropped head syndrome” Compartido por: ELA y MG Concomitante con 1er patrón Goldman: Cecil Medicine, 23rd ed.

17
Antecedentes Familiares
Patrones de herencia AD, AR, AR- LX y vertical materno (mitocondrial) IDENTIFICAR UN PATRÓN HEREDITARIO AYUDA AL DIAGNÓSTICO Y EN EL CONSEJO GENÉTICO Goldman: Cecil Medicine, 23rd ed.
 IDENTIFICAR UN PATRÓN HEREDITARIO AYUDA AL DIAGNÓSTICO Y EN EL CONSEJO GENÉTICO. Goldman: Cecil Medicine, 23rd ed.")
18
Medical Research Council of Great Britain (MRC) grading scale:
EXPLORACIÓN NEUROLÓGICA Graduar la fuerza muscular por grupos y bilateralmente Sentado extensión de la rodilla y flexión de cadera Posición prona flexión de la rodilla Decubito lateral abducción de la rodilla Medical Research Council of Great Britain (MRC) grading scale: 5 – Normal 4 – Movimiento activo contra la gravedad y la resistencia 3 – Movimiento activo contra la gravedad 2 – Movimiento activo sólo si se elimina la gravedad 1 – Se aprecia la contracción 0 – No hay contracción Medical Research Council. Aids to the Examination of the Peripheral Nervous System. Memorandum no. 45. London, Her Majesty's Stationery Office, 1981.
 grading scale: 5 – Normal. 4 – Movimiento activo contra la gravedad y la resistencia. 3 – Movimiento activo contra la gravedad. 2 – Movimiento activo sólo si se elimina la gravedad. 1 – Se aprecia la contracción. 0 – No hay contracción. Medical Research Council. Aids to the Examination of the Peripheral Nervous System. Memorandum no. 45. London, Her Majesty s Stationery Office,")
19
EXPLORACIÓN NEUROLÓGICA
Observar desempeño de las actividades de la vida diaria Sonrisa horizontal debilidad facial baja Imposibiliadad para cerrar los ojos completamente debilidad facial alta Ptosis Voz nasal debilidad palatina Goldman: Cecil Medicine, 23rd ed.

20
EXPLORACIÓN NEUROLÓGICA
MARCHA marcha miopática “de pato” con hiperlordosis (debilidad pélvica) Imposibilidad de caminar con los tobillos o puntas (debilidad distal) Levantarse de una silla o del piso (Signo de Gowers) Subir escaleras Goldman: Cecil Medicine, 23rd ed.
 Imposibilidad de caminar con los tobillos o puntas (debilidad distal) Levantarse de una silla o del piso (Signo de Gowers) Subir escaleras. Goldman: Cecil Medicine, 23rd ed.")
21
EXPLORACIÓN NEUROLÓGICA
Rigidez muscular probar la incapacidad para relajar el puño Percutir músculos con martillo de reflejos contracción persistente (región tenar y muñeca; grupo de músculos extensores de los dedos) Goldman: Cecil Medicine, 23rd ed.
 Goldman: Cecil Medicine, 23rd ed.")
22
LABORATORIO ENZIMAS SÉRICAS DE ORIGEN MUSCULAR
CPK Isoformas: MM (músculo esquelético); MB (músculo cardiaco); BB (cerebro) Su ausencia no excluye miopatía Puede elevarse x 5 en alteraciones neuromusculares Es el marcador más sensible de enfermedad muscular AST, ALT, DHL Elevadas en alteraciones hepáticas Su elevación obliga a medir la GGT y CPK Goldman: Cecil Medicine, 23rd ed.
; MB (músculo cardiaco); BB (cerebro) Su ausencia no excluye miopatía. Puede elevarse x 5 en alteraciones neuromusculares. Es el marcador más sensible de enfermedad muscular. AST, ALT, DHL. Elevadas en alteraciones hepáticas. Su elevación obliga a medir la GGT y CPK. Goldman: Cecil Medicine, 23rd ed.")
23
En reposo. Silencio eléctrico, salud de masa muscular. Si existe contracción es patológico. Fibrilación (denervatorio) y onda aguda positiva. En esfuerzo, potenciales de unidad motora (PUM), voltaje (200 microV a 2 mV), duración (5 a 18 mseg) y número de fases (bi o trifásicos). Miopatía: Actividad espontánea en reposo. Baja amplitud, voltaje Aminoff, M. Clinical electromyography. In: Electrodiagnosis in Clinical Neurology, Elsevier, Philadelphia p.233.
, voltaje (200 microV a 2 mV), duración (5 a 18 mseg) y número de fases (bi o trifásicos). Miopatía: Actividad espontánea en reposo. Baja amplitud, voltaje. Aminoff, M. Clinical electromyography. In: Electrodiagnosis in Clinical Neurology, Elsevier, Philadelphia p.233.")
24
Síntoma cardinal miopatías:
Debilidad muscular. Síntoma cardinal en enfermedades de placa neuromuscular: Fatigabilidad. Diferencia con alt neurogénica: Alt sensibilidad. Topografía déficit motor. Reflejos osteotendíneos. Fasciculaciones. Reflejo ideomuscular disminuido.

25
Se utiliza para tener el diagnóstico
Músculo moderadamente débil, no severamente (< 3/5) Miopático vs neuropático Neuropático atrofia por denervación con fibras angulares pequeñas, grupo de fibras atróficas y grupos de fibras histoquímicamente iguales Miopático núcleo central, hipertrofia de células pequeñas y grandes, degeneración y regeneración de fibras M Inflamatorio células inflamatorias mononucleares en endomisio y perimisio M mitocondrial fibras rojas rasgadas en la tinción de Gomori
 Miopático vs neuropático. Neuropático atrofia por denervación con fibras angulares pequeñas, grupo de fibras atróficas y grupos de fibras histoquímicamente iguales. Miopático núcleo central, hipertrofia de células pequeñas y grandes, degeneración y regeneración de fibras. M Inflamatorio células inflamatorias mononucleares en endomisio y perimisio. M mitocondrial fibras rojas rasgadas en la tinción de Gomori.")
26
Goldman: Cecil Medicine, 23rd ed.
Electrolitos séricos, pruebas endocrinas e inmunológicas específicas para establecer el diagnóstico Prueba de ejercicio del antebrazo Sospecha de miopatía metabólica que involucra la vía enzímpatica de la glucólisis Después de ejercicio vigoroso se mide los niveles séricos de lactato y amonio Deficiencia de fosforilasa sin elevación de lactato Análisis de orina Mioglobinuria (+ para sangre, sin eritrocitos) Goldman: Cecil Medicine, 23rd ed.
 Goldman: Cecil Medicine, 23rd ed.")
27
Goldman: Cecil Medicine, 23rd ed.
Microscopía electrónica Evalúa los componentes ultraestructurales No se requiere para hacer un diagnóstico patológico Se utiliza para el estudio de ciertas alteraciones en las microscopía de luz: Miopatías congénitas y mitocondriales Genética molecular Se utiliza para el diagnóstico y para conocer a los portadores Goldman: Cecil Medicine, 23rd ed.

28
Goldman: Cecil Medicine, 23rd ed.

29
Miopatías hereditarias
MIOPATIAS Clasificación Miopatías hereditarias Distrofias musculares Miopatía congénitas Monotonías y canalopatías Miopatías metabólicas

30
MIOPATIAS Clasificación Miopatías adquiridas Miopatías inflamatorias
Miopatías asociadas a enfermedades sistémicas Miopatías endocrinas Miopatías inducidas por drogas

31
MIOPATIAS CONGENITAS

32
M.C. Son un grupo de enfermedades hereditarias que se caracterizan por su comienzo congénito, curso generalmente benigno y presencia de rasgos morfológicos característicos. Neurol Clin N Am 21 (2003) 779–794
 779–794.")
34
Clasificacion modificada
1- Miopatías con alteración en la maduración o desarrollo muscular: a) Miopatía miotubular asociada al cromosoma X b) Desproporción congénita de tipos de fibras (21%) 2- Miopatías con anormalidades nucleares: a) Miopatías centronucleares (14%) 3- Miopatías con alteración de las proteínas miofibrilares y citoesqueléticas: a) Miopatías concores : - Enfermedad con concores centrales(16%) - Enfermedad con múltiples minicores (10%) b) Miopatía nemalínica. (20%)
 Miopatía miotubular asociada al cromosoma X. b) Desproporción congénita de tipos de fibras (21%) 2- Miopatías con anormalidades nucleares: a) Miopatías centronucleares (14%) 3- Miopatías con alteración de las proteínas miofibrilares y citoesqueléticas: a) Miopatías concores : - Enfermedad con concores centrales(16%) - Enfermedad con múltiples minicores (10%) b) Miopatía nemalínica. (20%)")
35
Comparten muchas características clínicas y patológicas, con severidad variable.
Inicio precoz. Casos severos: disminución o ausencia de movimientos fetales y partos complicados, seguidos de hipotonía y respiración poco efectiva. Incapacidad para alimentarse. Neurol Clin N Am 21 (2003) 779–794
 779–794.")
36
Más comunmente, el primer año presentan hipotonía, debilidad, infecciones respiratorias frecuentes.
Patrón difuso, predominio proximal. Niños pequeños y delgados. Paresia facial, voz nasal. Raro disfagia. Ptosis y oftalmoparesia puede ocurrir. Neurol Clin N Am 21 (2003) 779–794
 779–794.")
37
CK suele ser normal o solo ligeramente elevado.
EFS usualmente normal, a veces un patrón miopático o irritabilidad. No hay evidencia de defecto en unión neuromuscular. A veces la Bp muestra carácter distrófico como aumento de tejido conectivo endomisial. Suele ser necesario más de una biopsia. Inmunohistoquímica suele no ser necesaria para el diagnóstico. Sí ayudaría en la nemalínica con Ac contra alfa- actinina. Neurol Clin N Am 21 (2003) 779–794
 779–794.")
38
MIOPATIAS MITOCONDRIALES
Abordaje diagnostico

40
Determinación del acido láctico
Enzimas musculares EMG Biopsia muscular Microscopia electrónica Neuroimágenes Estudio molecular

41
Biopsia muscular La fibras rojo rasgadas son caracteristicas
(tincion de tricromico Gomori) y expresan un cambio morfologico secundario a una fosforilacion oxidativa defectuosa. Son fibras que se forman al cortar el músculo congelado se tiñen rojo en la periferia por acumulo de mitocondrias. Aparecen en mutaciones y deleciones del ADNmt que codifica RNAt no se ven en los que codifican proteinas estructurales (NARP y MILS)
 y expresan un cambio morfologico secundario a una fosforilacion oxidativa defectuosa. Son fibras que se forman al cortar el músculo congelado se tiñen rojo en la periferia por acumulo de mitocondrias. Aparecen en mutaciones y deleciones del ADNmt que codifica RNAt no se ven en los que codifican proteinas estructurales (NARP y MILS)")
42
Oftalmoplejia externa progresiva.
Encefalomiopatia mitocondrial con acidosis láctica y episodios de stroke-like (MELAS). Sindrome de Leigh de herencia materna (MILS). Encefalomiopatia neurogastrointestinal mitocondrial (MNGIE). Neuropatia ataxica sensitiva con disartria y oftalmoparesia (SANDO)
. Sindrome de Leigh de herencia materna (MILS). Encefalomiopatia neurogastrointestinal mitocondrial (MNGIE). Neuropatia ataxica sensitiva con disartria y oftalmoparesia (SANDO)")
43
MIOPATIAS INFLAMATORIAS
Dermatomiosistis Polimiositis Miositis en cuerpos de inclusion

44
Miopatias inflamatorias
Grupo heterogéneo de desórdenes caracterizados por inflamación del músculo esquelético, con daño de fibra y debilidad clínica. Existen dos categorías de miopatías. Idiopática. Infecciosa. Rheum Dis Clin N Am 32 (2006) 121–128
 121–128.")
45
DIAGNÓSTICO / CLASIFICACIÓN
1. Debilidad muscular (simétrica/proximal) 2. Incremento sérico de un componente muscular intracelular como enzimas (CPK) o mioglobina 3. EMG con anormlidades 4. Inflamación en la biopsia de músculo (1) muscle weakness (usually symmetric and proximal, sparing the muscles innervated by the cranial nerves); (2) an increase in the serum concentration of certain intracellular muscle components, such as enzymes (eg, creatinine kinase) or myoglobin; (3) an electromyogram showing abnormalities; and (4) inflammation seen on muscle biopsy. Classification criteria [3,4] suggest that a diagnosis of inflammatory myopathy is definite if all four of these elements are present, probable when three are present, and possible if two are present. CRITERIOS DE CLASIFICACIÓN: Diagnóstico = 4 elementos Probable = 3 elementos Posible = 2 elementos N Engl J Med 1975; 292(7):344– 7
 2. Incremento sérico de un componente muscular intracelular como enzimas (CPK) o mioglobina. 3. EMG con anormlidades. 4. Inflamación en la biopsia de músculo. (1) muscle weakness (usually symmetric and proximal, sparing the muscles innervated by the cranial nerves); (2) an increase in the serum concentration of certain intracellular muscle components, such as enzymes (eg, creatinine kinase) or myoglobin; (3) an electromyogram showing abnormalities; and (4) inflammation. seen on muscle biopsy. Classification criteria [3,4] suggest that a diagnosis of inflammatory myopathy is definite if all four of these elements are present, probable when three are present, and possible if two are present. CRITERIOS DE CLASIFICACIÓN: Diagnóstico = 4 elementos. Probable = 3 elementos. Posible = 2 elementos. N Engl J Med 1975; 292(7):344– 7.")
46
Dermatomiositis Cualquier edad M>H
Alteración microangiopatica mediada humoralmente. Depósitos vasculares de IgM, C3 que inducen un evento inmunológico primario, es la generación de Ac anti antígenos de la pared vascular los cuales evolucionan a daño isquémico de la fibra muscular. Su evolucion es de semanas a meses. Agudo (días) o progresivo. Rheum Dis Clin N Am 32 (2006) 121–128
 o progresivo. Rheum Dis Clin N Am 32 (2006) 121–128.")
47
Dermatomiositis Afección motora:
La debilidad puede ser precedida meses antes de: Fatiga, dolor y contractura muscular, disminución de la actividad y síndrome febril. Debilidad: Flexores de cuello. Cintura escapular y pélvica. Dificultad en levantarse del piso, de una silla, subir escaleras. Predomina la debilidad proximal Disfagia (33%), Afección cutánea. Rheum Dis Clin N Am 32 (2006) 121–128
, Afección cutánea. Rheum Dis Clin N Am 32 (2006) 121–128.")
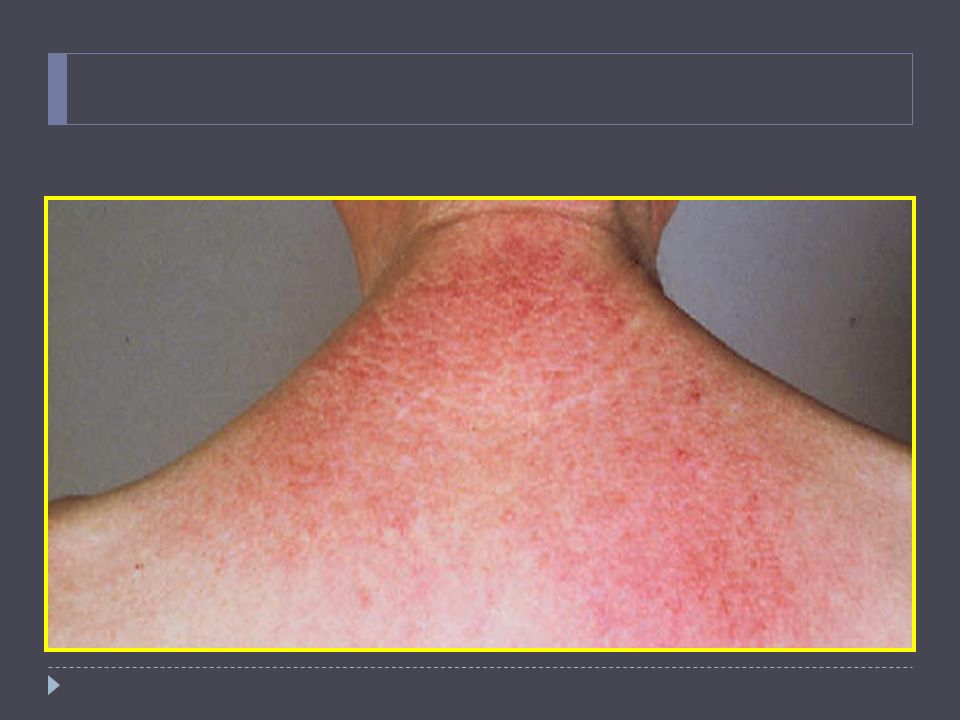
48
Dermatomiositis Lesiones papular, eritematosas y escamosas sobre nudillos (signo de Gottren). Eritema macular cara, cuello, cara anterior de tórax o sobre hombros y espalda (signo del chal). En niños aparece en 30 a 50% calcificaciones subcutáneas, en adultos no. Estas lesiones son dolorosas. Variante que desarrolla rash pero no debilidad: dermatomiositis amiopática. Rheum Dis Clin N Am 32 (2006) 121–128
. En niños aparece en 30 a 50% calcificaciones subcutáneas, en adultos no. Estas lesiones son dolorosas. Variante que desarrolla rash pero no debilidad: dermatomiositis amiopática. Rheum Dis Clin N Am 32 (2006) 121–128.")
51
Los niveles se correlacionan con la gravedad de la debilidad.
Evaluación incluye: Pruebas musculares. Electrocardiograma. Biopsia de músculo. Creatinin cinasa (CPK), marcador más seguro, sensible y específico de destrucción muscular, elevado en más del 90% de los pacientes. Niveles sobre 50 veces el valor normal. Los niveles se correlacionan con la gravedad de la debilidad. Rheum Dis Clin N Am 32 (2006) 121–128
, marcador más seguro, sensible y específico de destrucción muscular, elevado en más del 90% de los pacientes. Niveles sobre 50 veces el valor normal. Los niveles se correlacionan con la gravedad de la debilidad. Rheum Dis Clin N Am 32 (2006) 121–128.")
52
Lactato deshidrogenasa. Elevados, pero
Aldolasa. Mioglobina. Lactato deshidrogenasa Elevados, pero Aspartato aminotransferasa No aportan mayor Alanina aminotransferasa información. ANA + en 25 a 50%. Ac miositis específicos están asociados a HLA (limitados en clínica). Ac Jo – 1 + en pctes con enfermedad intersticial pulmonar. Rheum Dis Clin N Am 32 (2006) 121–128
. Ac Jo – 1 + en pctes con enfermedad intersticial pulmonar. Rheum Dis Clin N Am 32 (2006) 121–128.")
53
Electromiografia Ayuda en la determinación de que músculo biopsiar en casos leves. Aumento de la actividad espontánea. Potenciales de fibrilación. Positive sharp waves. Ocasionalmente descargas pseudomiotónicas o complejos repetitivos. Rheum Dis Clin N Am 32 (2006) 121–128
 121–128.")
54
Polimiositis Entre los 50 a 60 años. Raro en niños y cuando ocurre es dentro de un “síndrome de overlap”. Clínicamente se presenta similar a dermatomiositis, evolución de semanas a meses, con debilidad de flexores de cuello y proximal simétrico. Puede estar asociado a dolor. 30% disfagia. 10% enfermedad intersticial pulmonar (Ac anti Jo – 1). Poliartritis en 45%. Músculo cardíaco puede estar comprometido. Rheum Dis Clin N Am 32 (2006) 121–128
. Poliartritis en 45%. Músculo cardíaco puede estar comprometido. Rheum Dis Clin N Am 32 (2006) 121–128.")
55
CK sérica elevada 50 veces el valor normal.
ANA + en 30%. Ac miositis específicos +. Ac anti Jo – 1 en 20%. EMG: similar a dermatomiositis. Con patrón miopático de actividad espontánea aumentada, unidades motoras polifásicas pequeñas y reclutamiento temprano. Rheum Dis Clin N Am 32 (2006) 121–128
 121–128.")
56
DISTROFIAS MUSCULARES

57
Son miopatías genéticamente determinadas, usualmente causadas por un disturbio en la síntesis de una proteína estructural específica. Entidades que afectan principalmente al músculo estriado y que tienen en común un patrón distròfico de necrosis- regeneración característico en la biopsia muscular. Neurol Clin N Am 21 (2003) 795–816
 795–816.")
58
Componentes del cito esqueleto de la fibra muscular y del sarcolema:
La mayor parte de las distrofias tienen como causa anormalidades que involucran a las llamadas proteínas estructurales. Componentes del cito esqueleto de la fibra muscular y del sarcolema: Complejo de Glicoproteìnas asociadas a la distrofina. Neurol Clin N Am 21 (2003) 795–816
 795–816.")
59
OTROS ÓRGANOS IMPLICADOS
TIPO EDAD DE INICIO CUADRO CLÍNICO OTROS ÓRGANOS IMPLICADOS Duchenne < 5 AÑOS DEBILIDAD PROGRESIVA DE LA CINTURA PÉLVICA INCAPACIDAD PARA DEAMBULAR DESPUÉS DE LOS 12 AÑOS CIFOESCOLIOSIS PROGRESIVA INSUFICIENCIA RESPIRATORIA EN LA 2DA – 3RA DÉCADA DE LA VIDA CARDIOMIOPATÍA RETRASO MENTAL Becker 5 A LOS 25 AÑOS CAMINA DESPUÉS DE LOS 15 AÑOS INSUFICIENCIA RESPIRATORIA DESPUÉS DE LA 4TA DÉCADA CARDIOMIOPATIA Emery-Dreifuss NIÑEZ – ADULTOS CONTRACTURA DEL CODO Y DEL HÚMERO; DEBILDIAD PERINEAL Limb-Girdle DEBILDIAD PROGRESIVA DE LOS HOMBROS Y DE LA CINTURA PÉLVICA Neurol Clin N Am 21 (2003) 795–816
 795–816.")
60
Neurol Clin N Am 21 (2003) 795–816 TIPO EDAD DE INICIO CUADRO CLÍNICO
OTROS ÓRGANOS IMPLICADOS CONGÉNITO AL NACIMINETO O EN LOS 1ROS MESES HIPOTONÍA, CONTRACTURAS PROGRESIÓN A FALLA RESPIRATORIA ANORMALIDADES SNC Y OFTÁLMICAS FASCIO-ESCÁPULO-HUMERAL < 20 AÑOS DEBILDIAD LENTAMENTE PROGRESIVA DE LOS MÚSCULOS FACIALES, CINTURA ESCAPULAR Y DORSIFLEXIÓN DE LOS PIES SORDERA OCULOFARÍNGEO 5TA O 6TA DÉCADA DEBILIDAD LENTAMENTE PROGRESIVA DE LOS MÚSCULOS EXTRA-OCULARES, FARÍNGEOS Y DE LAS EXTREMIDADES ______ MIOTÓNICA 2DA DÉCADA DEFECTOS DE LA CONDUCCIÓN CARDIACA; ALTERACIONES MENTALES; CATARATAS; ATROFIA GONADAL Neurol Clin N Am 21 (2003) 795–816
 795–816.")
61
DISTROFIA DE DUCHENNE Y BECKER
Enfermedad de herencia recesiva ligada al cromosoma X. Dicho gen codifica la producciòn de distrofina. En la distrofia de Duchenne hay total carencia de distrofina. Frecuencia: de 1:3.500. Neurol Clin N Am 21 (2003) 795–816
 795–816.")
62
DISTROFIA DE DUCHENNE Y BECKER
Comienza entre los 2 y 4 años con retraso motor (40%), marcha anormal (30%), transtorno del lenguaje y el habla (8%). Excepcionalmente el inicio de los sìntomas es muy precoz, expresado por hipotonìa desde la lactancia temprana. Los signos caracterìsticos son: Debilidad de musculatura escàpulo-pèlvica, hipertrofia o seudohipertrofia gemelar, debilidad de los flexores del cuello. Neurol Clin N Am 21 (2003) 795–816
, marcha anormal (30%), transtorno del lenguaje y el habla (8%). Excepcionalmente el inicio de los sìntomas es muy precoz, expresado por hipotonìa desde la lactancia temprana. Los signos caracterìsticos son: Debilidad de musculatura escàpulo-pèlvica, hipertrofia o seudohipertrofia gemelar, debilidad de los flexores del cuello. Neurol Clin N Am 21 (2003) 795–816.")
63
DISTROFIA DE DUCHENNE Y BECKER
CI lìmite y ràpida progresiòn. Apariciòn posterior de retracciòn aquiliana, escoliosis y pèrdida de la ambulaciòn antes de los trece años. Signo de Gowers o maniobra de pararse trepando sobre si mismo es positivo. La expectativa de vida no sobrepasa la mitad de la tercera dècada. La muerte se debe a fallo respiratorio agudo o miocardiopatìa dilatada refractaria a tratamiento. Neurol Clin N Am 21 (2003) 795–816
 795–816.")
64
MANIOBRA DE GOWERS

65
DISTROFIA DE DUCHENNE Y BECKER
Estudios: CK elevada: Hasta mu/ml. Estudios de conducciòn nerviosa: Normales. EMG de aguja en reposo: Presencia de fibrilaciòn, ondas positivas y descargas de alta frecuencia. Patròn voluntario està compuesto por unidades motoras en general polifàsicas, breves y de baja amplitud. Neurol Clin N Am 21 (2003) 795–816
 795–816.")
66
DISTROFIA DE DUCHENNE Y BECKER
Biopsia muscular: Muestra un patròn distròfico con intensa fibrosis endomisial, fibras hipercontraìdas, diferentes grados de necrosis. Tècnicas de inmunohistoquìmica y de Western Blot: Se demuestra una ausencia total de distrofina. Neurol Clin N Am 21 (2003) 795–816
 795–816.")
67
DISTROFIA DUCHENNE MICROSCOPÌA

68
DISTROFIA DE DUCHENNE Y BECKER
DISTROFIA DE BECKER: Frecuencia: 1: varones. Sintomatologìa: Se inicia generalmente a los 5-15 años de edad, aunque los casos màs leves son de presentaciòn màs tardìa. Patròn de debilidad muscular similar a la de Duchenne. Con frecuencia existe intolerancia al ejercicio y mioglobinuria, y en ocasiones calambres. Neurol Clin N Am 21 (2003) 795–816
 795–816.")
69
DISTROFIA DE DUCHENNE Y BECKER
La clìnica es menos grave que en la forma Duchenne y los pacientes pierden la deambulaciòn unos 16 años despuès del comienzo de la enfermedad o incluso màs tardiamente. No es infrecuente la afectaciòn cardiaca, generalmente en forma de miocardiopatìa dilatada. EMG: Hallazgos menos acentuados que en la forma Duchenne. Neurol Clin N Am 21 (2003) 795–816
 795–816.")
70
DISTROFIA OCULO-FARINGEA

71
MIOPATIAS INDUCIDAS POR FARMACOS

72
Miopatías inducidas por medicamentos
¿ Incidencia ? Se cuenta dentro de las principales causas de mipatías Cuadro varíable: mialgia leve miopatía crónica + debilidad severa > 150 medicamentos asociados a rabdomiolisis Rheum Dis Clin North Am 1994 Nov;20(4):
:")
73
EtOH, cocaína, glucocorticoides, estatinas, antimalaricos y colchicina
Mecanismos: Toxicidad directa EtOH, cocaína, glucocorticoides, estatinas, antimalaricos y colchicina Inflamación inducida inmunológicamente D-penicilamina Daño muscular indirecto Isquemia por compresión y rabdomiolisis: opiacios y EtOH Hipokalemia (diuréticos) Estados hiperkinéticos (EtOH) Distonías (fenotiazinas) Muscle Nerve 2003 Feb;27(2):142-56
 Estados hiperkinéticos (EtOH) Distonías (fenotiazinas) Muscle Nerve 2003 Feb;27(2):")
Presentaciones similares